






Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne blodsykdommer
Hva er nytt?
Sist faglig oppdatert: 21.12.2023
Oppdateringen av Handlingsprogrammet Maligne blodsykdommer desember 2023 har vesentlige nyheter om:
- Kapittel 4 AML: Oppdatert med innføring av venetoklaks i kombinasjon med HMA etter vedtak i Nye metoder.
- Kapittel 5 ALL: Innføring av Blinatumomab i konsolodering ved CD19 posiitiv B-ALL som er MRD positive dag 29. Informomasjon om registering i WebCRF
- Kapittel 6 MPN: Innføring av Federatinib, oppdatering av prognosesystem og håndtering av splenomegali før transplantasjon.
- Kapittel 7 KLL: Oppdatert med informasjon om å ikke innføre venetoklaks i kombinasjon med rituksimab etter vedtak i Nye metoder.
- Kapittel 8 Myleomatose: Presisering om behandling med Belantamab og bispesifikke antisfoffer og CAR-T. Mindre oppdateringer om diagnostikk og prognostisering.
- Kapittel 12 MDS og KMML: Beskrivelse av diagnostiske kriterier i ICC og WHO 2023, Innføring av IPSS-Mol prognosesystem. Pressiseringer i behandling med melfalan, lenalidmoid og allogen stamcelletransplantsjon. Nytt kapittel om KMML. Kort om Vexas.
Innledning
Sist faglig oppdatert: 21.12.2023
Maligne blodsykdommer er en fellesbetegnelse på blodkreftsykdommer. Dette handlingsprogrammet omhandler blodkreftsykdommer som tradisjonelt behandles av spesialister i blodsykdommer (hematologi). Målgrupper for dette handlingsprogrammet er først og fremst leger og legespesialister innen indremedisin, hematologi, radiologi og andre medisinske fagområder som har ansvar for utredning, behandling og oppfølging av pasienter med maligne blodsykdommer. Det vil også være av interesse for andre legespesialister og faggrupper som møter pasientgruppen, som fastleger og sykepleiere og pasienter og pårørende.
Formålet med handlingsprogrammet er å sikre alle voksne norske pasienter med maligne blodsykdommer like muligheter for oppdatert diagnostikk og behandling basert på internasjonale, nasjonale og i størst mulig grad evidensbaserte behandlingsprinsipper. Handlingsprogrammet skal sikre korrekt klassifikasjon og subklassifikasjon av maligne blodsykdommer, spesielt i den gruppe der det er grunnlag for å gi behandling med tanke på å oppnå komplett remisjon og helbredelse.
Dette handlingsprogrammet omtaler diagnostikk og behandling for følgende sykdomsgrupper:
- Akutt myelogen leukemi
- Akutt lymfoblastisk leukemi/lymfoblastisk lymfom
- Myelodysplastisk syndrom
- Kronisk myelogen leukemi
- Myeloproliferative sykdommer
- Myelomatose
- Amyloidose
- Kronisk lymfatisk leukemi
- Morbus Waldenstrøm og andre indolente lymfomer.
Følgende kreftformer er ikke omhandlet i dette handlingsprogrammet:
- Lymfekreft, som i Norge for det meste behandles av spesialister i kreftsykdommer (onkologi)
Se: Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne lymfomer - Blodkreft hos barn
Se Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn
Maligne blodsykdommer er alvorlige sykdommer. Behandling og omsorg stiller store krav til helsepersonellet for å ivareta pasienters og pårørendes behov for informasjon, støtte og symptomlindring på en god måte. Vi viser til Nasjonalt handlingsprogram med retningslinjer for palliasjon i kreftomsorgen for omtale og anbefalinger om denne delen av omsorgen.
Når det gjelder seneffekter etter kreftbehandling, vises det til Rapport om seneffekter etter kreft.
Nye Metoder er et nasjonalt system for innføring og utfasing av metoder i spesialisthelsetjenesten. Det er de regionale helseforetakene som eier Nye metoder. Stortinget har vedtatt å lovfeste prioriteringskriteriene og at de regionale helseforetakene skal sørge for et felles system for å beslutte hvilke metoder som kan tilbys i spesialisthelsetjenesten. Handlingsprogrammene omtaler medikamenter og kombinasjoner på bakgrunn av evidens fra kliniske studier, og det informeresom finansiering. Et handlingsprogram kan ikke anbefale legemidler eller andre metoder som ikke har offentlig finansiering.
Metoder som er til vurdering skal som hovedregel ikke tas i bruk i norske sykehus. De regionale fagdirektørene har utarbeidet to prosedyrer for unntak fra bestemmelsen, en for grupper av pasienter og en for enkeltpasienter. Se Unntaksordning (nyemetoder.no)
Pakkeforløp
Sist faglig oppdatert: 21.12.2023
Pakkeforløp for myleomatose ble innført i helsetjenesten i 2015.
Pakkeforløp for kreft skal gi forutsigbarhet og trygghet for pasient og pårørende, og er et standard pasientforløp som beskriver organisering av utredning og behandling, kommunikasjon/dialog med pasient og pårørende samt ansvarsplassering og konkrete forløpstider. Pakkeforløpet starter når et helseforetak eller privat ideelt sykehus mottar en henvisning med begrunnet mistanke om kreft, eller når helseforetaket selv starter utredning med begrunnet mistanke om kreft.
Les mer om Pakkeforløp for myelomatose med tilhørende diagnoseveileder for inngang til pakkeforløp for kreft.
For pasienter med blodkreft som ikke har eget pakkeforløp, skal de ulike forløpstidene basere seg på medisinskfaglig vurdering i hvert enkelt tilfelle.
Pakkeforløp hjem for pasienter med kreft skal bidra til å sikre god struktur og logistikk i helsetjenesten, samt sikre trygghet og forutsigbarhet for personer som lever med kreft eller etter endt kreftbehandling. Les mer om Pakkeforløp hjem for pasienter med kreft.
Diagnostikk
Sist faglig oppdatert: 23.12.2021
Diagnostikk ved maligne blodsykdommer vil oftest være et samarbeid mellom kliniker, patolog, immunolog og genetiker. Diagnostikken følger WHO klassifikasjonen (Arber et al., 2016). Morfologisk undersøkelse av blod og beinmarg er sentral, og suppleres med data fra immunologiske, genetiske og molekylære undersøkelser.
I Norge vurderes tradisjonelt morfologi i utstrykspreparater fra blod og beinmarg av kliniker (hematolog) og i biopsier av patolog. Nært samarbeid mellom kliniker, patolog, immunolog og genetiker er nødvendig for å kunne integrere resultatene av klinikk og alle undersøkelser til en endelig diagnose i henhold til WHO-klassifikasjonen.
Pasientansvarlig kliniker har det endelige ansvar for å samle resultatene av alle de diagnostiske prøvene og tolke dem slik at det settes korrekt WHO-diagnose, og pasienten plasseres i rett prognostisk gruppe, spesielt der dette har betydning for behandlingsvalg. I fremtiden er det ønskelig å etablere hemato-onkologiske sentre som integrerer kliniske, morfologiske, immunologiske og genetiske funn.
Alle pasienter der intensiv behandling er aktuelt bør derfor som hovedregel henvises til universitetsklinikk for diagnostikk og planlegging av behandlingen. Større sykehus med erfaren hematolog og adekvat infrastruktur kan også påta seg diagnostiske oppgaver, men da alltid i nært samarbeid med universitetsklinikkene, både hva gjelder laboratorievurdering og klinisk evaluering.
Prøvetaking og transport
Sist faglig oppdatert: 23.12.2021
Diagnostisk prøvetaking bør om mulig gjøres tidlig i arbeidsuken (mandag til torsdag) slik at prøven kan nå laboratoriet før helg. Prøven bør om mulig tas før behandlingstart. Hvis dette ikke er mulig, og utsettelse av prøvetaking innebærer helserisiko for pasienten, må det forsøkes sikret diagnostisk materiale fra ubehandlet pasient til supplerende undersøkelser (immunfenotyping, cytogenetikk, molekylærpatologi) ved vitalfrysing med kryoproteksjon, evt fiksering før behandlingstart. Hvis mulighet for vitalfrysing mangler, oppbevares ufikserte prøver som må sendes ved romtemperatur inntil første arbeidsdag. Lufttørkede, ufikserte utstryk/rullepreparat er holdbare i mange dager. Gi eventuelt laboratoriet beskjed om at prøve er sendt; spesielt gjelder dette prøver som tas på fredag og/eller hvor prøvesvaret haster.
Relevant klinisk informasjon som det bør opplyses om på rekvisisjonen:
- Pasientdata, prøvetidspunkt, innleggelsesstatus, kjønn, rekvirent med navn og direkte telefonnummer / bemannet vakttelefon
- Type prøvemateriale
- Tentativ diagnose
- Hensikt med undersøkelsen: diagnostisk prøve, oppfølging, mistanke om hematologisk malignitet eller residiv
- Kort sykehistorie, langvarig medikamentell behandling, mistanke om tidligere kreftsykdom, evt annen eksponering som kan være relevant
- Kliniske data: perifere blodverdier, % blaster i blod, organ infiltrasjon
- Informer alltid om kjent smittefare: Hepatittvirus, HIV
Ved rekvirering av genetiske analyser:
- Behandling: kurativt siktemål? Behandling etter forskningsprotokoll?
- Hvilke andre genetiske analyser er rekvirert ved andre laboratorier?
- Ved oppfølgning: hvilke genetiske avvik som forelå tidligere
- Etter allogen stamcelletransplantasjon: hvilke genetiske avvik som forelå tidligere, donors kjønn.
- Dersom andre undersøkelser bekrefter eller endrer diagnosen bør laboratoriet informeres.
Klargjør før prøvetaking av blod og beinmarg prøverør/beholder med riktig antikoagulans/transportmedium/fikseringsvæske for:
- immunfenotyping: heparin(beinmarg), evt EDTA
- cytogenetikk: heparinisert prøve, evt i transportmedium
- molekylær patologi: EDTA eller heparin i transportmedium. For RNA baserte analyser (translokasjoner) ved HUS må PAX rør benyttes. Disse fås ved henvendelse til avdelingen.
Perifert blod
Bruk EDTA-blod til utstryk, immunfenotyping og genetiske undersøkelser med PCR. For cytogenetisk analyse benyttes heparin.
7 mL blod sikrer vanligvis nok materiale. Ved lave celletall bør man øke prøvevolumet. Ved akutt leukemi bør det om mulig vitalfryses separererte celler med DMSO for senere supplerende undersøkelser.
Beinmargsaspirat
Klargjør først prøverør og sprøyter fylt med riktig antikoagulans til ønskede prøver.
Til morfologi: Bruk fettfrie objektglass. Legg en liten dråpe blod/beinmarg på den ene enden av glasset. Man bør forsikre seg om at beinmargselementer er inkludert. Enkelte ganger kan det hjelpe å skylle ut sprøyten i EDTA for man aspirerer. Dette kan hindre at aspiratet klumper seg før utstryket lages.
Dra glasset til å lage utstryket (slepet utstryksglass) mot dråpen i en vinkel på 30 grader til det berører blod/ beinmargsdråpen. Blodet vil spre seg bak utstryksglasset ved hjelp av kapillarkraft, og man bør la det spre seg utover i glassets fulle bredde. Dra utstryksglasset lett og raskt nedover glasset slik at det dannes en fin hale. Lufttørk utstryket. Merk glasset med pasientens fulle navn.
Fiksering med metanol er å anbefale ved forsendelse. Utstryk til cytogenetiske analyser sendes ufiksert.
Til immunfenotyping og cytogenetikk: heparin (5000 IE/mL uten konserveringsmiddel) tilsatt sprøyten før aspirasjon (minimum 500 IE/mL aspirat).
Prøvevolum: 1–3 mL til hvert laboratorium (avhengig av leukocyttall og problemstilling). Antikoagulert prøve overføres til transportmedium (McCoy eller RPMI tilsendt fra laboratoriet).
Første aspirat har minst blodtilblandling og bør prioriteres for den viktigste analysen.
Beinmargsbiopsi
Beinmargsbiopsi bør være minst 1.5 cm lang, helst 2 cm, og fikseres snarest i formalin (4 %) eller i en zinkholdig fikseringsvæske (B+ væske). Unngå å klemme vevet ved biopsitaking. Biopsien fjernes fra nålen ved å føre sonden inn mot stikkretningen. Rullepreparater bør lages og kan brukes til FISH dersom det er dry tap. Kontakt lokalt patologilaboratorium for opplysninger om hvilken fiksering de foretrekker. Det er ønskelig at det sendes 2 ufargede +beinmargsutstryk sammen med benmargsbiopsien til patologilaboratoriet.
Det er viktig med tynne snitt (4 μm) for å oppnå best mulig morfologi. Som standard farges 3 snitt hvorav ett med hematoxylin/eosin, ett med Giemsa og ett med Gomori (sølvbasert farging av retikulinfibere).
Hvis flowcytometriske analyser ikke utføres, komplementeres morfologisk vurdering ofte med immunhistokjemiske farginger. Antistoffpanel er avhengig av morfologiske funn og kliniske opplysninger.
Ved dry tap kan en del av biopsien også holdes ufiksert for å lage en cellesuspensjon til flowcytometri og cytogenetikk. Hvis slik undersøkelse ønskes, må det på forhånd avtales. Slike biopsier legges i Ringer’s væske og sendes umiddelbart til laboratoriet.
Lymfeknutebiopsi
En biopsi bør vare så representativ som mulig. Ikke ta ut vev for diagnose kun fordi det er enklere å fjerne enn mer utbredt vevsaffeksjon på et vanskeligere tilgjengelig sted. Ta i stedet den biopsien som mest sannsynlig inneholder den mistenkte tumor. Ved generell perifer glandelsvulst er lyskebiopsi minst egnet for histologisk diagnostikk fordi lymfeknutene her oftere har reaktive forandringer enn i andre lokalisasjoner.
Ved mistanke om sykdom hvor flowcytometriske- og/eller genetiske undersøkelser er avgjørende (f.eks. lymfoblastisk lymfom/Burkitt lymfom), og det ikke er tumorceller i blod eller beinmarg, bør ferskt, ufiksert vev sendes direkte til et patologilaboratorium med spesialkompetanse innenfor hemato-onkologisk diagnostikk. Laboratoriet kan så fordele og videresende materialet til relevante undersøkelser.
Annet materiale
Spinalvæske, ascites, BAL og finnålsaspirat: Antikoagulans unødvendig, volum er avhengig av celletall (minst 1 mL). Bør analyseres innen 8 timer; rask celledød. Ved mistanke om CNS-affeksjon kan cytospinpreparater av spinalvæske farget med MGG og bedømt av erfaren hematolog gi diagnose. Preparatene kan evt. brukes til immuncytokjemi. Ved lave celletall (celletall <20 x 106/l) kan cytospinpreparater av cellesediment fra forsiktig sentrifugert spinalvæske (40 g, 5 min) være nyttig.
Forsendelse av prøver
Alt vev som kan nå laboratoriet samme dag, med unntak av beinmargsbiopsier, bør fortrinnsvis sendes ufiksert.
Vevsbiter legges i avkjølt Ringers væske i en mindre beholder, evt. kan benyttes fysiologisk saltvann eller annet transportmedium etter avtale med laboratoriet. Dersom materialet ikke forventes å nå fram til laboratoriet innen 30 minutter, bør beholderen med prøven holdes avkjølt på isbiter i egnet beholder (f.eks. termosflaske). NB! Tørris må ikke brukes. Maksimal transporttid 24 timer.
Dersom det er usikkert om en biopsi når laboratoriet i tide, er det hensiktsmessig å sikre noe av materialet for histologisk (innbefattet immunhistologisk) undersøkelse ved at deler av biopsien fikseres på 4 % bufret formaldehyd. Dersom ikke annet er mulig, kan hele biopsien sendes laboratoriet på 4 % bufret formaldehyd. I så tilfelle, bør kirurgen dele opp vevet i tynne skiver (ikke tykkere enn 3 mm) på langs for å sikre god fiksering.
Prøver sendes på raskeste måte (f.eks. med Postens Over Natten-service).
For prøvehåndtering og forsendelse av prøver for genetiske analyser se avsnittet "Prøvebehandling/transport" under kapittel Genetisk diagnostikk.
Morfologisk diagnostikk i blod og beinmargsaspirat
Sist faglig oppdatert: 23.12.2021
I Norge vurderes utstryk fra blod og beinmarg vanligvis av kliniker, oftest spesialist i hematologi. Diagnosen må kvalitetssikres ved vurdering ved sykehus med tilstrekkelig kompetanse (universitetssykehus og sykehus med spesialist i hematologi). Differensialtelling av beinmarg bør optimalt omfatte 500 kjerneholdige celler ved problemstilling hvor nøyaktige andeler er bestemmende for diagnose eller klassifikasjon. Det vises ellers til spesiallitteratur (Swerdlow et al., 2017).
Morfologisk diagnostikk i biopsi
Sist faglig oppdatert: 23.12.2021
Vurderes av patolog med erfaring i hematopatologi. Se spesiallitteratur (Swerdlow et al., 2017).
Genetisk diagnostikk
Sist faglig oppdatert: 23.12.2021
Hensikten med genetisk analyse ved malign blodsykdom er å undersøke forekomst av og karakterisere genetiske avvik i den maligne klon. Påvisning av kreftspesifikke avvik kan bidra til å skille malign fra benign tilstand, og bidra til å klassifisere neoplasien etter WHOs retningslinjer for diagnostikk (Swerdlow et al., 2017).
Mutasjonsstatus er assosiert med forventet behandlingsrespons og prognose og kan også påvirke valg av behandling. Påvist genetisk endring kan i mange tilfeller benyttes for vurdering av behandlingsrespons og minimal restsykdom (MRD). Ved mistanke om residiv kan genetisk analyse benyttes for å skille et residiv fra en behandlingsrelatert leukemi.
Undersøkelse for alle synlige kromosomale avvik kan gjøres ved bruk av G-båndsanalyse.
Målrettet undersøkelse på definerte avvik kan gjøres ved hjelp av fluorescens in situ hybridisering (FISH) eller ved PCR- baserte teknikker. PCR er rask og sensitiv (≥1x10–4). FISH derimot har lavere sensitivitet: 5–15 % avhengig av probe. FISH er mer anvendbar i tilfeller hvor samme gen kan inngå i flere ulike translokasjoner, eller hvor det er uvanlige bruddpunkt.
Mutasjoner i enkeltgener undersøkes med PCR. Hovedsakelig benyttes DNA, men analysen kan også utføres på RNA.
Deteksjon av tap eller tillegg av genmateriale og tap av heterozygositet kan gjøres med DNA matriser. Måling av genekspresjonsstatus i enkeltgener eller i hele transkriptomet kan gjøres ved hjelp av ekspresjonsmatriser. Disse matrisebaserte analysene inngår pt. ikke i noe diagnostisk tilbud i Norge.
Hver av de ulike klassene innen hematologiske maligniteter har sine spesifikke genetiske avvik. Det er derfor essensielt at det gis kortfattede kliniske opplysninger med klar problemstilling og dersom tentativ diagnose endres etter svar fra andre undersøkelser, bør dette videreformidles.
Bruken av genetiske analyser ved prognosesetting og behandlingsplanlegging ved maligne blodsykdommer er i meget rask utvikling, og dette handlingsprogrammet gir ikke komplett informasjon. Behovet for flere og mer kompliserte analyser og korte responstider er økende og kostnadskrevende.
Hvor skal prøven sendes
For genetiske analyser som beskrives i handlingsprogrammet: se tabell 3.1.
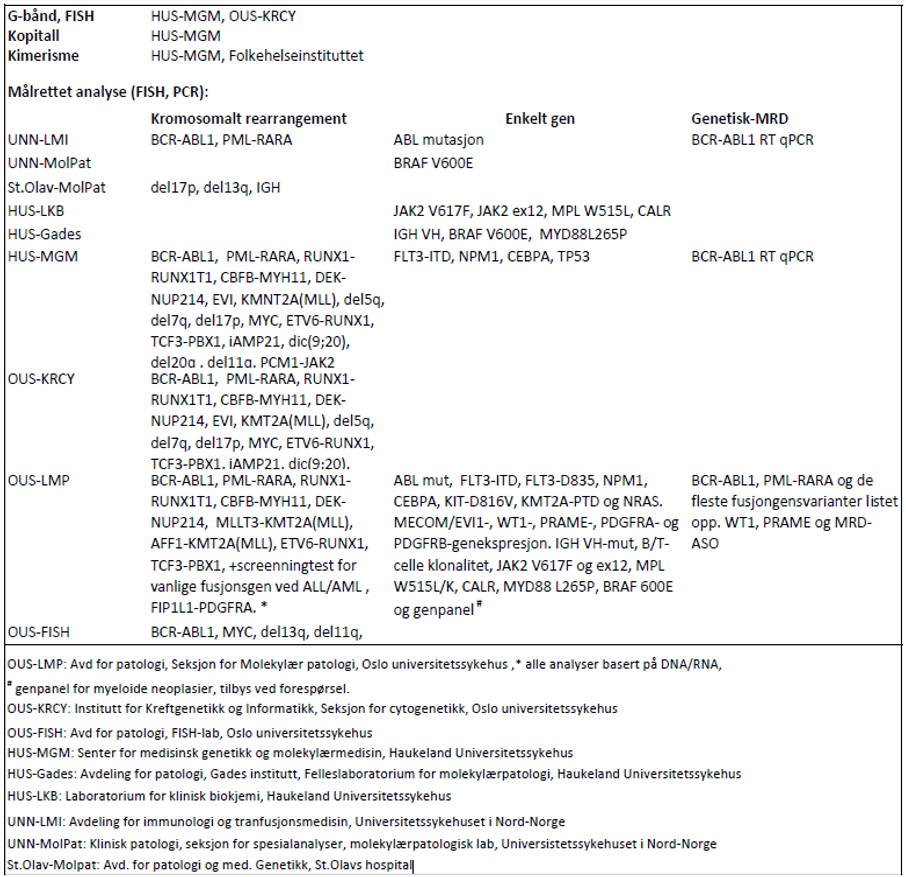
Laboratoriene har flere analyser i sitt tilbud enn det som fremkommer av tabellen og det henvises for fullstendig analyseoversikt og oppdaterte adresser til www.genetikkportalen.no. Her vil en også finne oversikt over andre aktuelle genetiske analyser som utføres i Norge.
Prøvebehandling/transport
For diagnostiske prøver bør prøven tas før behandlingen starter.
Cytogenetisk analyse (G-bånd og FISH) krever levende celler. Prøven må derfor alltid oppbevares ved romtemperatur; mellom 16–22 °C (unngå temperaturer under 4 °C og over 30 °C). Spesielle forbehold må tas på vinterstid for å unngå nedkjøling (isolasjon). Dersom transportfirma benyttes, må de gjøres oppmerksom på dette. Kortest mulig transporttid direkte til laboratoriet er nødvendig, maks 2 døgn. mRNA for RNA-basert diagnostikk er ustabilt og bør være på laboratoriet innen 24 timer. Ved HUS benyttes spesialrør (PAX) som stabiliserer RNA slik at den kan oppbevares inntil 72 timer i romtemperatur.
DNA-basert diagnostikk krever ikke viable celler. DNA er mer stabilt og tåler lenger transporttid.
Laboratoriene som utfører disse analysene har stengt i helgen og på helligdager. Cytogenetisk analyse krever dyrkning av celler og bør derfor tas mandag-torsdag. Med mindre annet er avtalt vil prøver som mottas fredag bli dyrket over helg, noe som ofte medfører færre metafaser som kan analyseres. Prøver for RNA-basert diagnostikk bør også tas mandag-torsdag dersom prøven må transporteres over lengre avstand, evt benytte spesialrør (PAX). Dersom behandling må påbegynnes utenfor åpningstiden bør det tas representative prøver før oppstart. Disse bør oppbevares i romtemperatur inntil de oversendes til laboratoriet. Hvis spesielle genetiske avvik kan utelukkes med FISH, vil dette kunne utføres på beinmargsutstryk.
Presis og korrekt merking av alle glass, rekvisisjon og forsendelsespapirer er viktig. Anfør om prøvesvaret haster. Gi eventuelt laboratoriet beskjed om at prøve er sendt, spesielt gjelder dette prøver som tas på fredag/før helligdager eller i tilfeller hvor prøvesvaret haster.
Prøvemateriale
For de fleste maligne sykdommer omfattet i dette handlingsprogrammet er det ønskelig med beinmargsmateriale, primært aspirat. Ved akutte leukemier kan perifert blod benyttes dersom det er påvist over 10 % blaster. Ved kroniske tilstander kan blod benyttes dersom aspirat er vanskelig å gjennomføre. Blod foretrekkes ved KLL. For MRD undersøkelser er det spesielt ønskelig med beinmarg, unntatt ved KML. Spinalvæske, ascites og finnålsaspirat ved mistanke om infiltrasjon i aktuelt organ kan også benyttes. Ved dry tap kan beinmargsbiopsi benyttes.
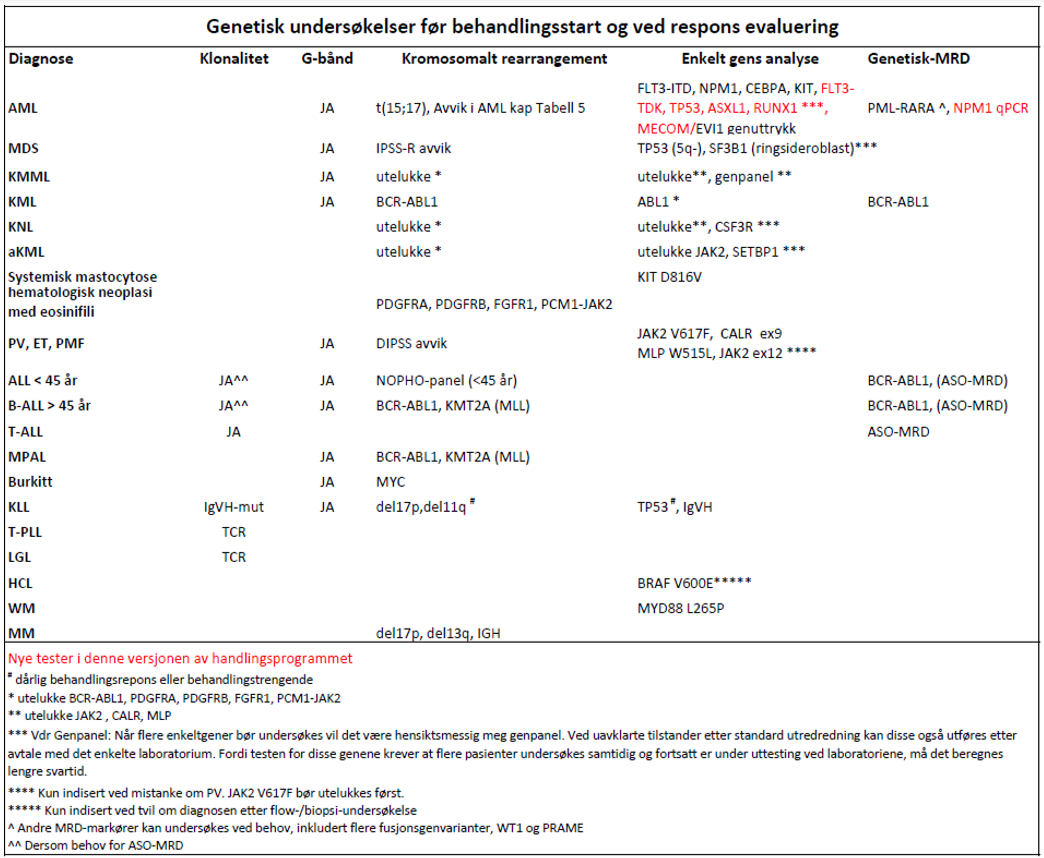
Problemstillinger for genetiske analyser
Normalt bør alltid blod- eller beinmargsutstryk vurderes før rekvirering for klarest mulig problemstilling. Dersom diagnose er usikker ved forsendelse må informasjon videreformidles til laboratoriet ved endringer eller bekreftelse av diagnose.
G-båndsanalyse benyttes for screening etter alle mulige kromosomale avvik. Metoden er tidkrevende og har begrenset sensitivitet. Den maligne klon må utgjøre så stor andel i prøvematerialet at den oppdages i minst 2–3 celler ved analyse av 20 metafaser.
Målrettet undersøkelse etter spesifikke genetiske avvik gjøres enten med bruk av FISH eller PCR. PCR er mest sensitiv. FISH har en fordel fordi et gen kan ha flere translokasjonspartnere, eller bruddstedet i genet kan variere mellom pasienter. Tap og tillegg av genområder eller kromosomer kan påvises med FISH.
For å benytte målrettet genetisk analyse for monitorering av behandlingsrespons (minimal restsykdom, MRD) må det foreligge et kjent avvik. Indikasjon for slik analyse foreligger dersom resultatet kan få praktisk konsekvens for behandlingen av pasienten, eller dersom undersøkelsen gjøres som ledd i en vitenskapelig undersøkelse. Det er stor forskjell i sensitivitet mellom de ulike genetiske metodene.
- Genetisk diagnostikk er indisert ved berettiget mistanke om akutt og kronisk leukemi, MDS, MPN og myelomatose (Haferlach et al., 2007).
Hvilke analyser kan/bør utføres
B-linje-ALL
Pasienter ≤45 år i NOPHO protokoll (vil endres ila 2018 ved oppstart av ALLtogether):
Analysen kan utføres på beinmarg, blod kan benyttes ved >30 % blaster
G-båndsanalyse. Ved normale funn analyseres minst 20 metafaser. Ved avvikende funn bør minst 10 metafaser analyseres (Hastings et al., 2013).
Målrettet analyse ved FISH eller RT-PCR: t(9;22) / BCR-ABL1, t(12;21) / ETV6-RUNX1, t(1;19) / TCF3-PBX1, dic(9;20), 11q23 / KMT2A (MLL) rearrangement, ic21amp.
Dersom RT-PCR er benyttet for KMT2A og ingen andre kromosomale rearrangement er påvist, bør FISH mot KMT2A utføres (nær 200 ulike KMT2A rearrangement er beskrevet og RT-PCR assayene er kun mot de vanligste).
Kopitallsanalyse anbefales dersom avvik ikke påvises ved de andre analysene, eller det er behov for verifikasjon av mistenkte ubalanserte avvik.
Hypodiploiditet måles ved flowcytometri og ved kopitallsanalyse.
Farmakogenetikk: Thiopurine methyltransferase (TPMT) genotyping av minimum G460A og A719G (EDTA blod, St. Olav, Avd for medisinsk genetikk, Medisinsk Genetisk laboratorium).
Pasienter ≥45 år:
G-båndsanalyse. Ved normale funn analyseres minst 20 metafaser.
Målrettet analyse: t(9;22) / BCR-ABL1, 11q23 / KMT2A (MLL) rearrangement.
Dersom RT-PCR er benyttet for KMT2A og ingen andre kromosomale rearrangement er påvist, bør FISH mot KMT2A utføres (nær 200 ulike KMT2A rearrangement er beskrevet og RT-PCR assayene er kun mot de vanligste).
Burkitt lymfom
Analysen bør utføres på beinmarg evt lymfeknute
G-båndsanalyse. Ved normale funn analyseres minst 20 metafaser (Hastings et al., 2013).
Målrettet analyse: 8q24 / MYC rearrangement
KLL
Analysen kan utføres på blod.
Ved diagnose:
- PCR: IGVH mutasjonsstatus
- Før behandling eller ved progresjon:
- G-båndsanalyse
- FISH minst 200 interfaser: del(17p13) / TP53
- Evt. del(11q22), del(6q), del(13q14.3) og trisomi 12
- PCR: TP53 (ekson 4–10) mutasjonsstatus
- del17p/TP53 bør gjentas ved hver behandlingslinje.
MDS
- Analysen bør utføres på beinmarg.
- G-båndsanalyse. Ved normale funn analyseres minst 20 metafaser (Hastings et al., 2013).
- Målrettet analyse: Dersom det ikke er 20 analyserbare metafaser av brukbar kvalitet, alternativt interfase FISH undersøkelse for -5/5q / EGR1, -7/7q /D7S486, +8 og del(20q) / D20S108, i(17q / TP53), 3q26 / EVI rearrangement
- Dersom ringsideroblaster: sekvensering av SF3B1 ekson 14–15 (Arber et al., 2016)
- MDS med isolert del(5q): sekvensering av TP53 mutasjon ekson 4–10 (Arber et al., 2016)
- Alle pasienter med MDS og KMML som er aktuelle for allogen stamcelletransplatasjon bør få utført neste generasjons sekvensering (NGS), myeloid panel (dvs. dypsekvensering ved hjelpe av TruSightMyeloid Sekvenserings panel)
AML
- Analysen kan utføres på beinmarg, blod kan benyttes ved >30 % blaster.
- G-båndsanalyse. Ved normale funn analyseres minst 20 metafaser (Hastings et al., 2013).
- Ved normal karyotype eller mislykket analyse bør målrettet analyse utføres på: t(15;17) / PML-RARA, t(8;21) / RUNX1-RUNX1T1, inv(16) / CBFB, t(9;22) / BCR-ABL1, 11q23 / KMT2A (MLL) rearrangement, 3q26 / MECOM (EVI) rearrangement, 5q- / EGR1, 7q-/-7 /D7S486, t(6;9) / DEK-NUP214, se tabell nedenfor.

Genpanel vil kunne benyttes der det er tilstrekkelig med deteksjon av 5 % av cellepopulasjonen og calling algoritmene klarer å identifisere mutasjonen. Denne analysen er kun tilgjengelig ved OUS-LMP.
For FLT3-ITD: semikvantiativ endepunkts PCR stoppet i eksponentiell fase separert på kapilærelektroforese. Ved påvist ITD beregnes ratio ut fra anbefaling fra ELN: areal under kurvene for FLT3-ITD (summert ved flere topper) over FLT3-normal allel. Det må gjøres oppmerksom på at HOVON beregner FLT3 ratio som areal FLT3-ITD (hovedtopp) / FLT3-ITD+FLT3-normal.
APL-mistanke: t(15;17) / PML-RARA, evt andre 17q21 / RARA rearrangement med RT-PCR. FISH kan benyttes dersom RNA ikke foreligger. MRD ved real time RT-PCR.
AML t(8;21): KIT ekson 8 med sekvensering og ekson 17 mutasjoner med sekvensering eller allelspesifikk analyse.
KML
Beinmarg sendes til cytogenetisk analyse («G-banding», karyotypering) og blod sendes til molekylær genetisk analyse (RT-PCR).
Cytogenetisk analyse bør utføres på beinmarg, men blod kan benyttes dersom >10 % blaster.
G-båndsanalyse ved diagnose: minst 3 celler med translokasjon som involverer kromosom 9q34 og 22q11, men helst 20 metafaser for å undersøke for klonale evolusjon (Hastings et al., 2013).
BCR-ABL1 fusjon skal verifiseres med målrettet analyse. RT-PCR basert analyse bør benyttes for å identifisere hvorvidt e13a2/e14a2 eller andre alternative transkript foreligger. Dette resultatet vil være av betydning for valg av analyse for å måle respons av behandling. FISH vil være aktuelt der vanlig t(9;22) ikke foreligger eller fusjon ikke er påvist med RT-PCR men kliniske symptomer stemmer med KML.
MPN (Polycytemia vera, Essensiell trombocytose, Primær Myelofibrose, Kronisk nøytrofil leukemi)
JAK2V617F mutasjon med allelspesifikk metode. Dersom negativ, vurderes mutasjonsanalyse for CALR (exon 9) og MPL W515L/K ved essensiell trombocytose og primær myelofibrose samt JAK2 ekson 12 ved polycytemia vera. Dersom de tre førstnevnte markørene er negative ved mistanke om primær myelofibrose, kan påvisning av andre hyppig forekommende mutasjoner (f.eks. ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1) støtte at det foreligger klonal sykdom (Arber et al., 2016).
For JAK2V617F er det anbefalt å ha en analytisk sensitivitet på minimum 1 %, men testen må ikke være for sensitiv da svært lave JAK2V617F mutasjon er funnet i friske individer (<0,1 %) (Lippert et al., 2009; Wang et al., 2008).
(Blod kan benyttes.)
For risikostratifisering ved primær myelofibrose: G-båndsanalyse. Sekvensering av ASXL1, EZH2, SRSF2 eller IDH1/2 kan vurderes.
Kronisk nøytrofil leukemi:
CSF3R T618I mutasjon eller annen aktiverende mutasjon av CSF3R (Arber et al., 2016).
Systemisk mastocytose
PCR: KITD816Vmutasjon med allelspesifikk metode med minimum 0,1 % sensitivitet (blod eller benmarg)
Myeloide og lymfoide neoplasmer assosiert med eosinofili
FISH/RT-PCR: PDGFRB, PDGFRA, FGFR1 rearrangement, PCM1-JAK2-fusjon (Arber et al., 2016) (analysen bør utføres på beinmarg)
MDS/MPN
JMML omtales ikke.
Det kreves fravær av BCR-ABL1 fusjonstranskript, rearrangering av PDGFRA/-B eller FGFR1 og PCM1-JAK2 (Arber et al., 2016).
Ved MDS/MPN med ringsideroblaster og trombocytose kreves fravær av (3;3)(q21;q26), inv(3)(q21q26) og del(5q). Det er da aktuelt å undersøke på SFRB1 mutasjon (Arber et al., 2016) og for mutasjon i JAK2 V617F, CALR eller MPL genene.
Dersom man ikke kan stille en sikker diagnose ved mistanke om KMML/aKML med tradisjonelle metoder (inkludert morfologi, flowcytometri og cytogenetikk), kan påvisning av de hyppigst assosierte mutasjonene støtte diagnosen (inkludert hhv. TET2, SRSF2, ASXL1 og SETBP1, samt SETBP1 og ETNK1) (Arber et al., 2016).
Myelomatose
Analysen bør utføres på CD138 celler isolert fra heparin-beinmarg. Standard oppsett tilstrekkelig for prognose og evt behandlingsvalg: FISH t(4;14)/IGH-FGFR3/MMSET, t(11;14)/ IGH-CCND1, del17p13/TP53. Se kapittel 8.7 Cytogenetikk. (St. Olav, Avdeling for patologi og medisinsk genetikk).
Hårcelleleukemi
Analysen bør utføres på benmarg.
BRAF V600E (kun indisert dersom man ikke kommer i mål immunfenotyping og morfologi)
Svarrapport
Rapporten skal inneholde følgende (Hastings et al., 2013):
- Hvilke analyser som er utført og resultatene på disse.
- Ved G-båndsanalyse skrives karyotype i henhold til ISCN nomenklatur 2016 (McGowan-Jordan et al., 2016).
- Antall celler som er analysert
- Ved sekvensering skrives sekvensvarianter ihht HGVS nomenklatur (15.11) (http://varnomen.hgvs.org/)
- Kort beskrivelse av klinisk relevante funn.
- Forhold mellom de enkelte funn og den kliniske problemstilling, evt andre mulig diagnoser (Heim et al., 2015).
- Ved normale resultater ved FISH/PCR analyse må deteksjonsgrense bemerkes (Bustin et al., 2010).
- Eventuelle begrensinger ved analysen eller nødvendige tilleggsanalyser bør bemerkes.
- Dersom det foreligger informasjon om forventet behandlingsrespons eller prognose bør dette også kommenteres, gjerne med prognosegruppe ihht til det gjeldende handlingsprogram.
Flowcytometrisk immunfenotyping
Sist faglig oppdatert: 23.12.2021
Hensikten med immunfenotyping ved leukemi og lymfom er å identifisere de(n) neoplastiske celletype(r). Mengde, linjetilhørighet og modningsnivå skal bestemmes, i tillegg til eventuelle unormale fenotyper som kan brukes til vurdering av minimal residual sykdom (MRD).
Det er essensielt at det gis kortfattede kliniske opplysninger med klar problemstilling slik at laboratoriet kan avgjøre hva som skal gjøres. Oppgi/rekvirer også leukocyttallet.
Prøvebehandling/transport
Immunfenotyping krever levende celler; prøven må derfor alltid oppbevares ved romtemperatur; mellom 16–22 °C (unngå temperaturer under 4 °C og over 30 °C). Kortest mulig transporttid direkte til laboratoriet er viktig. Presis og korrekt merking av alle glass, rekvisisjon og forsendelsespapirer er viktig. Gi eventuelt laboratoriet beskjed om at prøve er sendt.
Prøvemateriale
Vanligvis blod og/eller beinmargsmateriale. Ved akutt leukemi er beinmargsaspirat å foretrekke, men ved betydelig andel blaster i blod (ca >10 %) kan dette anvendes. Finnålsaspirat, spinalvæske, ascites ved mistanke om infiltrasjon i aktuelt organ.
Perifert blod: Antikoagulans; EDTA, (citrat går også), 3 mL vanligvis nok. Bør analyseres innen 2 døgn.
Beinmargsaspirat: Antikoagulans; heparin (5000 IE/mL uten konserveringsmiddel) tilsatt sprøyten før aspirasjon (min 500 IE/mL aspirat) eller citrat. Bør analyseres raskest mulig, helst innen 1 døgn. (Spesielt AML celler forringes betydelig i løpet av første døgn).
Prøvevolum: 1–3 mL (avhengig av leukocyttall og problemstilling).
Spinalvæske: Antikoagulans unødvendig, volum er avhengig av celletall (minst 1 mL). Celledød skjer meget raskt (>90 % etter 1 time). Spinalvæsken bør tilsettes stabiliserende transportmedium ved prøvetaking (for spesialmedium; kontakt laboratorium).
Ascites: BAL og finnålsaspirat: Antikoagulans; som for spinalvæske.
Biopsier: Antikoagulans unødvendig. Tilsettes 1 mL isoton saltvann.
Problemstillinger for immunfenotyping
Meningsfylte resultater fra immunfenotyping forutsetter en klar problemstilling. Et utstryk bør normalt alltid være vurdert før immunfenotyping, med mindre indikasjonen er sterk. Immunfenotyping er indisert ved (Bene et al., 2011; Davis et al., 2007):
Klar indikasjon:
- Utredning av akutt leukemi
- Mistanke om CNS-affeksjon ved akutt leukemi
- Utredning av kronisk lymfoproliferativ lidelse
- Utredning av lymfom
- Mistanke om paroksysmal nokturnal hemoglobinuri (perifert blod)
- MRD ved ALL.
Immunfenotyping kan være nyttig:
Diagnosen vil i disse tilfellene ofte baseres på andre kriterier enn immunfenotyping, og undersøkelsen bør da utgå når den ikke er diagnostisk nødvendig. Behandlende lege bør vurdere i hvert enkelt tilfelle om immunfenotyping er indisert.
- Mistanke om blastkrise ved KML; diagnosen blastkrise stilles uten immunfenotyping, men etter at diagnosen eventuelt er stilt kan det utføres immunfenotyping for å avgjøre om det er en myeloid eller lymfatisk blastkrise.
- Kvantitering av CD34+ celler i perifert blod ved diagnose av myelofibrose og myelodysplasi dersom man mener dette er avgjørende for den prognostiske vurderingen. Ellers har immunfenotyping ingen plass ved kontroll av myelofibrose og myelodysplasi.
- Myelomatose; kun ved tvil om diagnosen
- Mistanke om Richters transformasjon, ellers har immunfenotyping ingen plass ved kontroll av KLL
- Diagnose av KMML
- Diagnose av KML (ikke nødvendig ved typiske funn ellers)
- MDS
- MRD ved AML etter 2. konsolideringskur og før allogen stamcelletransplantasjon
Resultater
Linjetilhørighet kan en sjelden gang være vanskelig å bestemme da det ikke finnes helt spesifikke markører. Det henvises til gjeldende WHO klassifikasjon for kriterier for tilkjenning av linjetilhørighet og også for modning. Det er viktig å vite at andel «blaster» ved immunfenotyping ikke nødvendigvis vil være identisk med blaster observert ved mikroskopi.
Mengden av en populasjon angis som prosent av viable leukocytter eller som andel av alle viable kjerneholdige celler. Vær oppmerksom på at grad av blodtilblanding vil influere resultatet av immunfenotyping sammenliknet med utstryk (med mindre man bruker materiale fra samme uttrekk til begge undersøkelsene).
Populasjoners immunfenotypiske normalitet vurderes ut fra om de har aberrant ekspresjon av linjemarkører de normalt ikke skal ha (f.eks. CD7 på myeloide celler), eller om de har for lite/for mye av en markør (f.eks. for mye CD10 på B precursor celler), eller om de uttrykker markører som normalt ikke skal være til stede samtidig (f.eks. CD10++ og CD20 på B celler).
Rapport:
Den immunfenotypiske rapport bør blant annet inneholde (Bene et al., 2011; Davis et al., 2007):
- Hva som er analysert
- Celletetthet og kvaliteten (viabilitet) av materialet
- Hvilke populasjoner som er vurdert og % andelen av abnormale
- Med hvilke markører og intensiteten av disse:
- Positiv, negativ og delvis positiv i forhold til en intern negativ populasjon
- Sterkt, normalt, svakt, heterogent i forhold til normalpopulasjonen
- Antigen som kan bli nyttige til eventuell MRD nevnes
- De viktigste funnene oppsummeres og en eventuell konklusjon gis
- WHO-basert klassifikasjon skal tilstrebes. Evt. avvik fra gjeldende WHO-klassifikasjon bør bemerkes.
Behandlingsrespons, minimal restsykdom og monitorering av sykdomsprogresjon
Sist faglig oppdatert: 23.12.2021
ALL
Leukemispesifikke markører med immunfenotypisk eller molekylærgenetisk metode ved diagnosetidspunkt benyttes til å bestemme MRD markør. B-ALL brukes vanligvis flowcytometri, og ved T-ALL PCR av T-celle reseptor. Dersom man ikke finner MRD markør ved anbefalt metode, brukes alternativ metode (PCR for B-ALL og væskestrømcytometri for T-ALL). MRD vurderes som negativ ved verdier mindre enn 10–3 (<0,1 % leukemiceller) for flow og mindre enn 10–4 ved PCR.
Beinmargsaspirat benyttes. For Flow cytometri lyseres røde blodceller og for kvantivativ PCR isoleres mononukleære celler. Se NOPHO ALL-2008 MRD-flow and MRD-PCR guidelines.
Kontroll etter dag 29 og dag 80–100 etter oppstart av behandling. Hos eldre kan man tilpasse individuelt, men man vil vanligvis nøye seg med MRD dag 29, dag 79 og etter 12 måneder.
Hos MRD-negative høyrisk pasienter med donor som ikke transplanteres, bør MRD måles hver 3. måned det første året etter avsluttet konsolidering
Ved mistanke om residiv bør det sendes ny prøve til flow cytometri og genetisk analyse. Målet med genetisk analyse er å kunne skille mellom residiv og behandlingsrelatert leukemi, samt ved residiv undersøke for genetisk evolusjon. Alle genetiske analyser utført ved diagnose er derfor ikke nødvendig.
AML
Leukemispesifikke markører med immunfenotypisk (flow-MRD) eller molekylærgenetisk metode ved diagnosetidspunkt benyttes til å bestemme MRD markør. Genetisk MRD ved AML er ansett som sandard hos pasienter med APL og pasienter med NPM1 mutasjon. Hvordan flow-MRD skal benyttes hos pasienter uten NPM1 eller APL er fortsatt ikke avklart, men flere studier indikerer at flow MRD kan benyttes til å vurdere indikasjon for allogen stamcelletransplantsjon, spesielt hos pasienter med intermediær risiko.
MRD anbefales nå rutinemessig i følgende situasjoner:
- Ved APL: behandlingsrespons måles ved PML-RARA ved kvantitativ RT-PCR etter 3. kur for lavrisk og 2 uker etter avsluttet konsolidering ved høyrisk. For høyrisk utføres MRD også hver 3. måned i 2 år etter avsluttet behandling.
- pm1 mutasjon uten andre mutasjoner: kvantitativ PCR etter kur, så videre hver 6. uke i 2 år
- Hos pasienter med core binding factor leukemi, hver 6. uke i 2år
- Flow-MRD etter kur nr 2 og før allogen stamcelletransplantasjon som en uavhengig prognostisk markør.
Funn som indikerer suboptimal respons/ residivskal alltid verifiseres med ny prøve til flow cytometri og genetisk analyse.
KML
Behandlingsrespons måles både ved cytogenetisk (beinmarg) og molekylær genetisk (blod) analyse.
Cytogenetisk analyse: G-banding av minimum 20 metafaser. Kontroll utføres etter 3. og 6. måneder. behandling, senere hver 6. måned til stabil CCgR er oppnådd, deretter årlig til stabil MMR. Dersom stabil MMR, behøves ikke cytogenetisk undersøkelse såframt ikke den molekylære monitoreringen viser tegn til tap av respons. Analysen bør utføres på beinmarg. Dersom BCR-ABL1 fusjonen skyldes ett kryptisk rearrangement bør kontrollanalysene utføres med FISH utfra FISH mønster påvist i diagnoseprøven.
Molekylær genetisk analyse: Fusjonstranskriptet påvist ved diagnose bestemmer hvilket assay som benyttes for kvantitativ RT-PCR. Ved alternative transkript: se tabell 3.1 for hvilket laboratorium som tilbyr analysen.
Da testens sensitivitet bestemmes utfra kopitallet av referansegenet bør det foreligge minst 10 000 ABL kopier eller 24 000 GUS kopier per replikat. Testens sensitivitet bør fremgå av svaret. Svaret bør beregnes ut fra summen av verdiene og besvares i internasjonal standard og respons kurve bør være tilgjengelig (Cross et al., 2015).
Kontroll utføres hver 3. måned til stabil MMR, deretter hver 6. måned. Ved «advarsel» bør ABL1 mutasjonsanalyse utføres. Ved seponering månedlig de første 6 måneder, hver 2. måned de neste 6 og deretter hver 3. måned. Ved rebehandling kontrolleres de hver 3. måned.
Myelomatose
Per i dag er det uklart hvorledes MRD-analyser bør brukes i praksis. MRD ved flow eller sekvensering som et relevant endepunkt i kliniske studier. I vanlig klinisk praksis utenom studier er det imidlertid fullgodt per i dag å bruke de tradisjonelle responskriteriene.
Etter allogen stamcelletransplantasjon
Analysen bør utføres på beinmarg.
Transplantasjon etter BCR-ABL1 positiv akutt lymfatisk leukemi som ikke behandles med imatinib, måles BCR/ABL i blod hver 3.–4. uke og i marg hver 6.–8. uke det første året.
Andre maligniteter monitoreres med kimerisme undersøkelse.
(ved Folkehelseinstituttet, Område for rettsmedisinske fag og Senter for medisinsk genetikk og molekylærmedisin, Haukeland sykehus). Laboratoriet må ha mottatt prøve fra donor og pasient før transplantasjon.
Akutt myelogen leukemi (AML)
Utredning og diagnostikk ved akutt myelogen leukemi
Sist faglig oppdatert: 23.12.2021
For sikker diagnostikk av akutt myelogen leukemi (AML) kreves:
- Morfologisk undersøkelse av beinmargsutstryk etter MGG farging.
- Flowcytometrisk undersøkelse av beinmarg, alternativt cytokjemisk farging.
- For diagnosen akutt promyelocyttleukemi kreves funn at translokasjonen t(15;17)(q24.1;q21.2) eller funksjonsprodukt PML-RARα påvises.
Hos pasienter der man vurderer å gi AML rettet behandling (både intensiv induksjonsbehandling men også leukemi-stabiliserende kjemoterapi) kreves:
- Cytogenetisk undersøkelse av beinmargceller
- Molekylærgenetisk undersøkelser av leukemicellene.
- Hos pasienter der intensiv kjemoterapi eller allogen stamcelletransplantasjon anbefales NGS-analyse av myeloide fokusgener (myeloid panel)
- AML diagnostikk skal skje ved eller i samarbeid med universitetssykehus.
- Man benytter European Leukemia Nets som grunnlag for den prognostiske vurderingen av pasienter med AML.
- For alle pasienter som er kandidat for intensiv kjemoterapibehandling bør pasient, deres søsken og foreldre vevstypes (evidensgrad D).
Behandling av akutt myelogen leukemi (ikke-APL-varianter)
Sist faglig oppdatert: 23.12.2021
Man tilrår følgende for intensiv induksjonsbehandling:
- Ved induksjonsbehandling hos pasienter opp til 65 år skal gis daunorubicin ≥60 mg/m2/d. Anbefalingen er daunorubicin 90 mg/m2 daglig i 3 dager eller idarubicin 12 mg/m2 daglig i 3 dager, begge kombinert med cytarabin 200 mg/m2 kroppsoverflate/døgn som kontinuerlig døgninfusjon i 7 døgn (evidensgrad A).
- Induksjonsbehandling hos pasienter i alderen 66-80 år etter individuell vurdering er daunorubicin 60 mg/m2/dag i 3 dager og cytarabin 200 mg/m2/dag i 7 døgn (evidensgrad A).
- Induksjonsbehandling bør startes så snart som mulig og senest 5 døgn etter at leukemi-diagnosen er stilt (evidensgrad C).
- Hos pasienter med FLT3 mutasjon anbefales tillegg av midostaurin fra dag 8 til 21 ved induksjonsbehandling, se under spesielle situasjoner for detaljer om konsolideringsbehandling og etter allogen stamcelletransplantasjon.
Pasienter i første remisjon tilbys konsolidering i samsvar med ett av følgende anbefalte regimer:
- Pasienter inntil 65 år kan behandles i samsvar med HOVON-SAKK protokoll som hos noen pasienter vil inkludere autolog stamcelletransplantasjon (evidensgrad B).
- Alternativt kan pasienter inntil 60 år behandles med høydose cytarabin 3 g/m2 to ganger daglig dag 1, 3 og 5 i gjentatte kurer med om lag 4 ukers mellomrom, inntil 4 kurer (evidensgrad B).
- Alternativt kan pasienter over 6065 år tilbys et regime med enten (i) en konsolideringskur basert på cytarabin intermediær dose; eller (ii) gjentatte kurer med lavdosert daunorubicin og subkutan cytarabin (evidensgrad B).
- For pasienter der man mener at ytterligere konsolideringsbehandling med intensiv kjemoterapi eller allogen stamcelletransplantsjon ikke er aktuelt grunnet stor risiko for prosedyre-relatert død kan man gi azacytidine månedlig i ett år (evidensgrad C).
Allogen stamcelletransplantasjon (SCT) i CR1:
- Høy residivrisiko uten vesentlig komorbiditet: aktuelt med familie- (evidensgrad A) eller ubeslektet giver (evidensgrad B).
- Intermediær residivrisiko under 40 år: aktuelt med familiegiver (evidensgrad B). Med ubeslektet giver aktuelt ved genomisk HLA-A, B, C, DRB1, DQB1-identitiet (10/10 match) og lav risiko for prosedyrerelatert mortalitet (evidensgrad C).
- Intermediær residivrisiko 40 eller eldre år: kan være aktuelt med familie- eller 10/10 match ubeslektet giver, men nøye vurdering av gevinst vs risiko sammen med pasienten (evidensgrad C).
Analyse av Minimal residual disease (MRD, minimal restsykdom) kan forbedre risikovurderingen i forhold til om man skal tilby konsolidering med allogen stamcelletransplantasjon:
- Foreløpig anbefales kun å bruke MRD analyse hos pasienter med NPM1 mutasjon tilskal brukes hos pasienter med har NPM1 mutasjon som eneste påviste genetiske avvik som konkret (evidensgrad C).
- MRD med immunfenotypisk (flow-MRD) er en sterk uavhengig prediktor for residiv etter allogen stamcelletransplantsjon og anbefales å undersøkes etter 2. induksjonskur og før allogen stamcelletransplantsjon (evidensgrad C).
- Flow-MRD anbefales foreløpig ikke å inkludere som egen prognostisk markør som indikasjon for allogen stamcelletransplantasjon for pasienter med intermediær eller høy risiko AML.
Anbefalinger ved spesielle situasjonene:
- For pasienter med FLT3 positiv AML anbefales tillegg av midostaurin dag 8 til 21 ved konsoliderende behandling (evidensgrad B).
- For pasienter med FLT3 positiv AML som ikke behandles med allogen stamcelletransplantsjon anbefales ett års vedlikeholdsbehandling med midostaurin (evidensgrad B).
- For pasienter med FLT3 positiv AML som behandles med allogen stamcelletransplantasjon anbefales vedlokeholdsbehandling med sorafinib (evidensgrad B).
- Ved AML i svangerskapet skal behandling skje ved Regionsykehus (evidensgrad D).
- Ekstramedullær sykdom diagnostiseres og behandles i samsvar med generelle retningslinjer for AML behandling. Man kan vurdere lokal strålebehandling mot eventuell restlesjon (evidensgrad D).
- Hos pasienter med isolert ekstramedullær sykdom uten påvisbare genetiske avvik, tidligere MDS der man ikke kan tilordne til en risikogruppe etter ELN anbefales ikke allogen stamcelletransplantasjon, men kjemoterapi som konsoliderende behandling i CR1.
- CNS affeksjon: Man gir ikke rutinemessig profylakse, behandling skjer ved intratekal cytostatika; initialt cytarabin monoterapi og, ved utilfredsstillende respons, kombinasjonsbehandling (evidensgrad D).
- Hyperleukocytose: Intensiv induksjonsbehandling startes snarest mulig, leukaferese vurderes kun dersom man har symptomgivende leukostase. Hos pasienter som ikke skal ha intensiv kjemoterapi benyttes subcutan cytarabin som blastreduserende behandling (evidensgrad D).
Anbefalinger for behandling av primært refraktær sykdom og residiv av AML opp til 65–75 år:
- Alternativer for behandling av residiv under pågående behandling eller innen 9–12 måneder etter avsluttet behandling:
- Induksjonsbehandling med alternativt kombinasjonsregime av cytostatika (evidensgrad C). eller behandling med venetoclax/azacitidine (evidensgrad D).
- Alternativt kan vurderes allogen stamcelletransplantasjon uten at man har oppnådd komplett remisjon (evidensgrad C).
- Allogen stamcelletransplantasjon som konsolidering etter oppnådd remisjon (evidensgrad A).
- Palliativ behandling (evidensgrad D).
Residiv mer enn 9–12 måneder etter avsluttet behandling
- Induksjonsbehandling med anthracyklin/cytarabin (evidensgrad C)
- Venetoclax/azacitidin kan forsøkes ved manglendre respons etter induksjonsbehandling og fortsatt indikasjon for allogen stamcelletransplantasjon (evidensgrad D)
- Allogen SCT som konsolidering ved oppnådd remisjon (evidensgrad B)
- Alternativt vurdere allogen stamcelletransplantasjon uten oppnådd remisjon (evidensgrad B).
Palliativ behandling (evidensgrad D).
Anbefaling, behandling av pasienter som ikke kan få intensiv terapi:
Alle pasienter skal ha behandling med transfusjoner og antibiotika. Azacitidine eller cytarabin kan benyttes som sykdom-stabiliserende terapi; azacitidin bør foretrekkes hos pasienter med høyrisiko cytogenetikk og multilineær dysplasi. Decitabine kan være et alternativ for pasienter med høyrisiko karyotype eller TP53 mutasjon (evidensgrad C).
Håndtering av pasienter med akutt promyelocyt leukemi (APL)
Sist faglig oppdatert: 23.12.2021
Anbefaling for den umiddelbare håndtering av pasienter med mistenkt APL:
- Alle norske sykehus skal ha all-trans-retin syre (Vesanoid/ATRA) tabletter tilgjengelig for umiddelbar bruk.
- Hos pasienter med akutt leukemi og der man ut fra klinisk bilde, morfologiske funn eller væskestrømcytometrisk undersøkelse mistenker APL, skal man umiddelbart starte med ATRA 45 mg/m2 fordelt på to doser daglig. Pasientene skal snarest mulig overføres sykehus med tilstrekkelig kompetanse for endelig rask diagnostisk avklaring og eventuell start av APL behandling.
- Ved klinisk mistanke om APL kontinueres behandling med ATRA frem til mistanken er endelig avkreftet med genetiske analyser.
- Ved mistanke om APL gjøres genetisk utredning som for andre former for AML. Imidlertid må genetisk laboratorium varsles slik at svar på analyse av PML-RARA fusjonstranskript kan foreligge snarest mulig, det vil si innen et par dager.
Generelle retningslinjer for behandling av APL:
- Hos pasienter med nydiagnostisert APL skal man gi transfusjoner av trombocytt- og fibrinogenkonsentrat for å holde trombocytter over 30-50x109/l og fibrinogen over 1,5 g/l fram til koagulopatien ikke lenger er til stede (evidensgrad A).
- Pasienter som etter oppstart av ATRA og eller arsenikk-trioksid (ATO) utvikler tegn på differesieringssyndrom skal umiddelbart starte behandling med intravenøs dexametason 10 mg morgen og kveld (evidensgrad A).
- Vi anbefaler generelt ikke profylaktisk behandling med dexamethason, men det kan overveies hos pasienter med nyresvikt og/ eller leukocytter over 10 x 109/l (evidensgrad C)
- Pasienter med APL klassifiseres enten som lavrisiko-sykdom ved LPK <10 x 109/l eller som høyrisikosykdom ved LPK på diagnosetidspunktet ≥ 10x109/l (evidensgrad A).
Behandling av pasienter med lavrisiko APL:
- Pasienter med lavrisiko-sykdom behandles uansett alder med ATRA og ATO (evidensgrad A).
- Hos pasienter med lavrisikosykdom er det ikke aktuelt med vedlikeholdsbehandling ut over 4 konsolideringskurer med ATRA og ATO. Det er kun anbefalt 1 MRD prøve som tas etter 3. konsolideringskur (evidensgrad B).
Behandling av pasienter med høyrisiko APL:
- Behandling av pasienter med høyrisikosykdom anbefales ATRA og ATO med tillegg av antracyclin (evidensgrad C).
- Pasienter med høyrisiko sykdom skal følges med MRD status i benmarg hver 3. måned i 1 år og hver 6. måneder i år 2 etter avsluttet behandling (evidensgrad A).
Behandling av APL tilbakefall:
- Pasienter under 70-75 år som etter primærbehandlingen opplever residiv (molekylærgenetisk eller morfologisk) bør få reinduksjon og påfølgende stamcelletransplantasjon (evidensgrad B).
Pasienter som etter induksjonsbehandlingen oppnår MRD negativ status, er kandidat for autolog stamcelletransplantasjon, mens pasienter som er vedvarende MRD positive bør vurderes for allogen stamcelletransplantasjon så sant det ikke foreligger kontraindikasjon (evidensgrad B).
Definisjoner
Sist faglig oppdatert: 23.12.2021
Følgende definisjoner er nødvendige å kjenne til når man skal diagnostisere og behandle AML (Döhner et al., 2017).
Hematologisk AML tilbakefall: Blaster i beinmarg ≥5 %, eller igjen påvisbare blaster i perifert blod, eller ekstramedullær sykdom. Dersom man har blaster mellom 5 og 10 % i beinmargen må funnet verifiseres i kontrollprøve.
Induksjonskur: Intensiv kur gitt for å oppnå komplett remisjon.
Komplett remisjon (CR): Blaster i beinmarg <5 %, ingen blaster med Auer-staver og ingen blaster i perifert blod, ingen ekstramedullær sykdom, nøytrofile i perifert blod ≥1.0 x 109/L, platetall i perifert blod ≥100 x 109/L.
Komplett remisjon uten MRD: Komplett remisjon som definert ovenfor og i tillegg ikke påvist minimal rest-sykdom (MRD, minimal residual disease)
Komplett remisjon med ufullstendig/inkomplett rekonstitusjon (CRi). Oppfyller kriteriene for komplett remisjon bortsett fra at man har vedvarende nøytropeni i perifert blod <1.0 x 109/L eller vedvarende trombocytopeni <100 x 109/L.
Konsolideringskur: Intensiv kur gitt etter oppnådd komplett remisjon for å utrydde restsykdom.
Morfologisk AML-fri status: Blaster i beinmarg <5 %, ingen blaster med Auer-staver, ingen ekstramedullær sykdom (ingen krav til hematologisk rekonstitusjon).
MRD (minimal residual disease, minimal restsykdom): Sykdomsceller som bare kan påvises med svært følsomme metoder og som ikke kan påvises ved morfologiske undersøkelser.
Primært refraktær sykdom: Ingen komplett remisjon eller komplett remisjon med ufullstendig regenerasjon etter 2 sykluser med intensiv induksjonsbehandling.
Progredierende sykdom: Definisjonene er ikke allment akseptert, men de er foreslått av ELN og kan fungere som en rettesnor i forbindelse med AML-stabiliserende behandling:
- Beinmargskriterier: (i) >50 % økning av blaster i beinmarg og dersom <30 % blaster i utgangspunktet kreves en stigning svarende til minst 15 %; alternativt vedvarende blaster i beinmargen over 70 % i minst 3 måneder; og (ii) uten at man samtidig har dobling av nøytrofile eller stigning av blodplater til >50 x 109/L. Eller:
- Kriterier i perifert blod aleine: >50 % økning av blaster konsentrasjon i perifert blod til >25 x 109/L uten at dette skyldes differensieringssyndrom. Eller:
- Nytilkommen ekstramedullær sykdom.
Epidemiologi og patogenese
Sist faglig oppdatert: 23.12.2021
Det diagnostiseres årlig om lag 150 nye tilfeller av akutt myelogene leukemi (AML) i Norge. Svært få av disse er barn, median alder ved diagnosetidspunktet er snaut 70 år og sykdommen er noe hyppigere hos menn enn hos kvinner (Döhner et al., 2017). Årsaken til AML hos den enkelte pasient er oftest ukjent (Döhner et al., 2017). Tidligere stråleterapi og cytostatikabehandling øker risikoen for AML gjennom deres genotoksiske effekter, og i disse tilfellene kan man ofte påvise bestemte genetiske avvik i AML cellene som indikerer en slik årsakssammenheng. En beskrivelse av det genomiske landskap ved AML og hvordan dette danner et grunnlag for klassifisering og prognosevurdering, er nylig publisert (Papaemmanuil et al., 2016). Man regner med at 10 til 15 % av pasientene har genetisk predisposisjon til AML. Grunnet forbedret diagnostikk med NGS-analyse av myeloide fokus gener fanges denne pasientkategorien opp i større grad enn tidligere.
I forhold til de behandlingsmessige konsekvenser kan AML inndeles i to hovedgrupper, akutt promyelocytleukemi (APL) og ikke-APL. APL har spesielle genetiske avvik og krever spesiell håndtering i forhold til andre AML typer. Så sant noe annet ikke er spesielt presisert bruker vi derfor i disse retningslinjene begrepet AML for ikke-APL variantene og omtaler disse sammen, mens APL omtales for seg i egne kapitler.
Man vil også presisere at de anbefalinger og retningslinjer som gis i denne gjennomgangen bare må oppfattes som veiledende i forhold til den individuelle vurdering som kreves for hver enkelt pasient.
Diagnostikk og klassifisering av AML
Sist faglig oppdatert: 23.12.2021
Definisjon. Det generelle kriteriet for diagnosen AML er minst 20 % myeloblaster av totalt antall kjerneholdige celler i beinmargen. Det er 5 unntak der diagnosen kan stilles uten at kravet om 20 % blaster må oppfylles; dette er ved de genetiske avvikene t(8;21) (q22;q22), t(16;16) (p13.1;q22), inv(16) (p13.1q22), t(15;17) (q22;q12) (akutt promyelocytleukemi) og i to spesielle tilfeller der man har overvekt i margen av erytroide forstadier (se tabell 4.1) (Arber et al., 2016).
Morfologisk vurdering i May-Grünwald-Giemsa farget beinmargsutstryk. Det eneste entydige morfologiske funn for å skille AML og akutt lymfoblastisk leukemi (ALL) ved mikroskopi av May-Grünwald-Giemsa (MGG) farget beinmargsutstryk er påvisning av Auer-staver i cytoplasma; dette finnes bare ved AML men kun hos et mindretall av pasientene. Akutt erytroleukemi ble tidligere inndelt i to undergrupper. Denne inndelingen er nå forlatt i revidert utgave av WHO-klassifikasjonen. Tabell 4.1 angir diagnose når kjerneholdige erytrocytforstadier utgjør minst 50 % av kjerneholdige beinmargceller (tilpasset fra Aber og medarbeidere (Arber et al., 2016)).
Tabell 4.1: Morfologisk diagnostikk
Morfologisk diagnostikk når andel kjerneholdige erytrocytforstadier utgjør mer enn 50 % av kjerneholdige beinmargsceller (grå markering indikerer at kravet om minst 20 % blaster av alle kjerneholdige celler ikke kreves oppfylt).
Diagnose | Kjerneholdige røde | Blaster av alle kjerneholdige | Tidligere cellegiftterapi | WHO genetisk endring* | Myelodysplasirelaterte endringer |
|---|---|---|---|---|---|
Terapi-relatert myeloid neoplasi** | ≥50 % | Ingen krav | Ja | Ingen krav | Ingen krav |
AML with recurrent genetic abnormality | ≥50 % | ≥20 % | Nei | Ja | Ingen krav |
AML with myelodysplasia-related changes | ≥50 % | ≥20 % | Nei | Nei | Minst 50 % dysplastiske celler i 2 linjer |
AML, NOS | ≥50 % | ≥20 % | Nei | Nei | Nei; oppfyller ikke kravene ovenfor |
MDS | ≥50 % | <20 % av ikke-erytroide celler | Nei | Nei | Ingen krav |
AML NOS, acute erytroid leukemia | >80 % umodne erytroide forstadier men ≥30 % pro-erytroblaster | <20 % | Nei | Nei | Ingen krav |
* Tilfeller av AML med t(8;21) (q22;q22); Runx1-RUNX1T1, t(16;16) (p13.1;q22), inv(16) (p13.1q22);CBFB-MYH11 eller APL med t(15;17) (q22;q12) påvises i sjeldne tilfeller og blir da avgjørende for klassifiseringen.
** WHO 2016 angitt Terapi-relatert myeloid neoplasi som en ny hovedgruppe. Morfologisk sett kan de undergrupperes i terapirelatert myelodysplastisk syndrom (t-MDS), MDS/myeloproliferativ neoplasi (t‑MDS/MPN) og AML (t-AML). De har likevel skilt terapi-relatert myeloid neoplasi ut som en egen hovedgruppe som inkluderer alle disse tre undergruppene.
Beinmargsbiopsi er sjelden nødvendig for å stille diagnosen AML, men det er helt nødvendig når man ikke får aspirert beinmarg på grunn av fibrose eller svært høy cellularitet. Dersom man mistenker erytroleukemi må man utelukke vitamin B12 og folatmangel. Diagnosen akutt megakaryoblast-leukemi krever immunfenotyping og som regel også benmargsbiopsi.
Cytokjemiske og biokjemiske undersøkelser av AML cellene. Disse undersøkelsene er stort sett erstattet av immunfenotyping. Diamin-benzidin peroxydase (DAB) farging er et supplement til MGG fargning; et alternativ er farging med Sudansvart. Esterasefarging kan være nyttig for diagnosen monocytt-myelomonocytt AML.
Immunfenotyping av AML celler med væskestrømcytometri. Flowcytometri benyttes til å avgjøre blastenes linjetilhørighet og differensieringsgrad; det regnes som en rutinediagnostikk hos alle AML-pasienter. Diagnosen akutt megakaryoblastleukemi krever immunfenotyping med væskestrømcytometri og/eller immunhistokjemi (CD61 og/eller CD41 positive blaster). Denne undergruppen kan ofte ha fibrose i margen, slik at man ikke får adekvat aspirat og diagnosen må baseres på funn ved biopsi og flowcytometri.
Tabell 4.2: Immunfenotyping for diagnose av AML (Döhner et al., 2017)
Differensiering | Markører |
|---|---|
Prekursorceller | CD34, CD117, HLA-DR (CD38, CD123 og/eller CD133 kan også inkluderes som stamcellemarkører men er ikke avgjørende for diagnose). |
Granulocytt | CD65, cytoplasmatisk myeloperoksidase (cMPO) Celler som er differensiert i retning granulocytter kan i varierende grad beholde uttrykk av CD13 og CD33. CD11b og CD15 kan være nyttig mens CD16 bare er uttrykt på modne granulocytter. Fravær av MPO men med andre myeloide markører skiller AML med minimal differensiering fra AML uten modning. |
Monocytt | CD14, CD36, CD34. Celler med monocytoid differensiering vil ofte ha bevart uttrykk av CD13 og CD33. Analyse av CD64 og CD11b kan gi tilleggsinformasjon spesielt for promonocytter |
Megakaryocytt | CD41 (glykoprotein IIb/IIIa), CD42 (glykoprotein 1b), CD61 (glykoprotein IIIa) |
Erytroide markører | CD36, CD235a (glykophorin A) |
Akutt udifferensiert leukemi (AUL) og akutt leukemi med blandet fenotype (mixed phenotype acute leukemia, MPAL). En sjelden gang uttrykker blastene ingen linjemarkører (akutt udifferensiert leukemi, AUL), eller de uttrykker samtidig markører som vanligvis regnes som linjespesifikke for myeloide og T/B lymfoide celler, eller de har flere subpopulasjoner som uttrykker ulik linjetilhørighet (mixed phenotype acute leukemia, MPAL). Disse klassifiseres i undergruppen akutte leukemier med usikker linjetilhørighet (tabell 4.3) og bør sannsynligvis ha induksjonsbehandling som ved ALL (Arber et al., 2016; Charles et al., 2017; Wolach et al., 2015, 2017). Noen pasienter med MPAL har t(9;22) (q34.1;q11.2) eller BCR-ABL1 translokasjonen; fordi det for dette genetiske avviket finnes målrettet behandling bør man alltid ved MPAL undersøke for dette avviket.
Diagnosen MPAL kan være vanskelig, ytterligere detaljer gis også i European Group for the Immunological Classification of Leukaemias (EGIL-klassifikasjonen) (Pomerantz et al., 2016).
Tabell 4.3: Akutt leukemi med usikker linjetilhørighet
Acute undiffentiated leukemia (AUL) eller Mixed phenotype acute leukemia (MPAL) (Arber et al., 2016)
DIAGNOSTISKE KRAV som må oppfylles for å angi mer enn en linjetilhørighet for en |
|---|
Myeloid (i) Myeloperoksydase (flowcytometri, histokjemi, cytokjemi); eller (ii) monocytdifferensiering definert som minst to av følgende: Ikke-spesifikk esterase, CDC11c, CD14, CD64 eller lysozym |
T celle linje (i) Cytoplasmatisk CD3 (flowcytometri med antistoff mot CD3 epsilon kjeden); eller (ii) Overflateuttrykk av CD3 (sjelden) |
B celle linje (i) Sterkt uttrykt CD19 og i tillegg sterkt uttrykk av minst en av følgende tre markører: CD79a, cytoplasmatisk CD22, CD10; eller (ii) Svakt uttrykk av CD19 og sterkt uttrykk av minst 2 av CD79a, cytoplasmatisk CD22, CD10 |
KLASSIFISERING |
Akutt udifferensiert leukemi (AUL): Ofte uttrykk av HLA-DR, CD34 og/eller CD38 (inntil en membranmarkør uttrykt for hver linje, men ikke uttrykk av myeloperoksydase, cytoplasmatisk CD3 eller B-celle markører som cCD22, cCD79a eller sterkt uttrykt CD19) |
MPAL med t(9;22)(q34.1;q11.2); BCR-ABL1 og ikke tidligere kjent KML. Som regel en dobbelt-positiv blastpopulasjon |
MPAL med t(v;11q23.3); KMT2A rearrangert; ofte en myeloid og en lymfoid cellepopulasjon men kan også ha dobbeltpositiv enkeltpopulasjon. |
MPAL: B/myeloid, NOS (ofte to subpopulasjoner) |
MPAL: T/myeloid, NOS (ofte dobbelt-positiv blastpopulasjon) |
MPAL, ikke ellers klassifisert |
Cytogenetiske og molekylærgenetiske undersøkelser. De genetiske analysene benyttes for prognostiske vurderinger, men de er avgjørende for diagnosen i de tilfellene der man fraviker det diagnostiske kravet om minst 20 % myeloblaster i beinmargen inv(16); t(16;16), t(8;21), t(15;17). Man bør hos alle pasienter sikre seg den genetiske diagnostikk som kreves for ELNs risikoklassifisering, se også kapittel 3 Diagnostikk.
Ved karyotyping påvises kromosomforandringer (det vil si samme forandring i to eller flere mitoser fra beinmargceller) hos vel halvparten av pasientene med AML. Visse cytogenetiske forandringer er assosiert med spesielle morfologiske forandringer. Inversjon av kromosom 16, inv(16)(p13.1q22), eller translokasjon mellom kromosom 16p og 16q, t(16;16) (p13.1;q22), ses ved myelomonocyttleukemi med samtidig eosinofili i beinmargen. Molekylærgenetisk finner man her fusjonsgenet CBF beta/MYH11. En del pasienter med AML med modning har translokasjonen t(8;21)(q22;q22) som gir fusjonsgenet RUNX1-RUNX1T1, også kalt AML1-ETO. Hos disse pasientene ses ofte en tydelig oppklaring i Golgi-sonen i MGG-preparat av beinmargsutstryk. Ved APL finner man hos de fleste pasienter translokasjonen t(15;17)(q24.1;q21.2) som gir fusjonsgenet PML-RARA (se eget kapittel).
Molekylærgenetiske analyser, NGS-analyse av myeloide fokus gener (myeloid panel), gir tilleggsinformasjon ved normal karyotype og pasienter med andre påvisbare genetiske avvik. Så langt er det kun mutasjoner i gene for NPM1, FLT3, TP53, ASXL1, RUNX1 som påvirker prognose. Mutasjon i NPM1 og FLT3 fanges opp av standard diagnostikk, mens mutasjoner i TP53, ASXL1 og RUNX1 fanges opp av myeloid panel. Myeloid panel gir også informasjon om det kan foreligger kimbanemutasjon. Myeloid panel anbefales til alle pasienter som gjennomgår intensiv kjemoterapi og/eller er kandidat for allogen stamcelletransplantasjon.
Genetisk predisposisjon for AML
Man tror at ca. 10 til 15 % av pasienter har en genetisk predisposisjon for AML. NGS diagnostikk gjør at kimbanemutasjoner i større grad en tidligere identifiseres. AML med genetisk predisposisjon deles inn i følgende grupper.
- Familiær AML uten andre kliniske manifestasjoner (eks CEBPAα mutasjon)
- Familiær AML med sykdom i angre organer (eks GATA2 mutasjon)
- Familiær AML med konstitusjonell trombocytopeni (eks. RUNX1 mutasjon, ANKRD26 mutasjons og ETV6 mutasjons)
- Medfødte benargssviktsyndromer (eks. Fanconis anemi og telomersykdommer)
- MECCOM syndrom
Ved mistanke om genetisk predisposisjon henvises pasienten/familien til genetisk utredning. For en detaljert oversikt over de forskjellige tilstandene vises til nordiske retningslinjer for diagnostikk og oppfølging.
Generelt anbefales at følgende familier/pasienter henvises genetisk utredning.
- Familier/ personer med MDS eller AML yngre enn 50 år ved diagnose og andre kliniske manifestasjoner assosiert med arvelig tilstand.
- Familier med to første eller andregradsslektninger med AML eller MDS, en yngre enn 50 år ved diagnose.
- Familier med en person med AML eller MDS med en første eller andregradsslektninger med annen cancer, en yngre enn 50 år ved diagnose.
- Første eller andregradsslektninger med MDS/AML eller vedvarende trombocytopeni eller andre kliniske manifestasjoner assosiert med hereditær AML.
- Personer med AML eller MDS med påvist forandringer av kromosom 7 yngre enn 50 ved diagnose
Tabell 4.4 er en oversikt over anbefalt oppfølging av friske personer/pasienter med påvist genetisk predisposisjon til AML. Det kan være aktuelt å tilby allogen stamcelle transplantasjon allerede ved påvist klonal evolusjon før utvikling av MDS eller AML. Ved funn av ervervet mutasjoner ved NGS analyse/ klonal evolusjon anbefales å henvise pasientene til transsplantasjonssenter for utredning av allogen stamcelletransplantsjon.
Tabell 4.4: Råd for oppfølging av pasienter med genetisk predisposisjon for MDS og AML, tilpasset fra nordiske retningslinjer
Analyse | Ved diagnose | Oppfølging |
|---|---|---|
Hematogram | Ja | Hver 6. måned |
Benmargsaspirat/biopsi | Ja | Ved mistanke om progresjon til MDS/AML |
NGS Myeloid panel | Ja | Hver 12. måned |
Annen diagnostikk. Bildediagnostikk er bare unntaksvis av betydning ved AML. Ved mistanke om leukemi utenfor beinmarg/blod er CT eller MR undersøkelser aktuelle; påviste lesjoner bør undersøkes cytologisk/histologisk og genetisk. Hos pasienter med myeloid sarkom kan det være aktuelt å følge pasienter med PET-CT for å senere å kunne evaluere respons etter behandling. CNS-manifestasjoner er svært uvanlig på diagnosetidspunktet, men kan forkomme seinere i forløpet spesielt ved monocytt/monoblast varianter av AML. Pasienter med CNS- symptomer skal derfor spinalpunkteres, men først når det ikke lenger er sirkulerende blaster i blodet. Det utføres da MGG-farging av cytospinpreparat samt væskestrømcytometri av spinalvæsken.
Anbefaling for diagnostikk av AML:
- For sikker diagnostikk av akutt myelogen leukemi kreves:
- Morfologisk undersøkelse av beinmargsutstryk etter MGG fargning.
- Væskestrømcytometrisk undersøkelse av beinmarg, alternativt cytokjemisk farging.
- Hos pasienter der man vurderer å gi AML-rettet behandling (både intensiv induksjonsbehandling men også leukemi-stabiliserende kjemoterapi) kreves i tillegg:
- Cytogenetisk undersøkelse av beinmargceller.
- Molekylærgenetisk undersøkelser av leukemicellene inkludert NGS-analyse av myeloide fokus gener (myeloid panel).
- Denne integrerte diagnostikken krever nært samarbeid mellom kliniker og laboratorium med spesialkompetanse, og vurderingene skal derfor skje ved universitetsklinikk eller i samarbeid med universitetssykehus.
Tabell 4.5: Klassifisering av akutte leukemier
Akutt myelogen leukemi (AML) og akutt leukemi med usikker linjetilhørighet.
WHO 2016 (Arber et al., 2016).
Acute myeloid leukaemia with recurrent genetic abnormalities |
AML with t(8;21)(q22;q22.1); RUNX-RUNX1T1 |
AML with inv(16)(p 13.1q22) or t(16;16)(p 13.1;q22); CBFB-MYH11 |
APL with PML-RARA |
AML with t(9;11)(p 21.3;q23.3); MLLT3-KMT2A |
AML with t(6;9)(p 23;q34.1); DEK-NUP214 |
AML with inv(3)(q21.3;q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM |
AML (megakaryoblastic) with t(1;22)(p 13.3;q13.3); RBM15-MKL1 |
AML with mutated NPM1 |
AML with biallelic mutations of CEBPA |
Provisional entity: AML with BCR-ABL1 |
Provisional entity: AML with mutated RUNX1 |
Acute myeloid leukaemia with myelodysplasia-related changes |
Therapy-related myeloid neoplasms |
Acute myeloid leukaemia, not otherwise specified (NOS) |
AML with minimal differentiation |
AML without maturation |
AML with maturation |
Acute myelomonocytic leukaemia |
Acute monoblastic and monocytic leukemia |
Acute erytroid leukaemia – Pure erythroid type |
Acute megakaryoblastic leukemia |
Acute basophilic leukaemia |
Acute panmyelosis with myelofibrosis |
Myeloid sarcoma |
Myeloid proliferations related to Down syndrome |
Transient abnormal myelopoiesis |
Myeloid leukaemia associated with Down syndrome |
Blastic plasmacytoid dendritic cell neoplasms |
Acute leukemias of ambiguous lineage (se også tabell 4.5) |
Acute undifferentiated leukemia (AUL) |
Mixed phenotype acute leukemia (MPAL) with t(9;22); BCR-ABL1 |
MPAL with t(v;11q23.3); KMT2A rearraged |
MPAL, B/myeloid, NOS |
MPAL, T/myeloid, NOS |
Prognostisk vurdering av pasienter med AML
Sist faglig oppdatert: 23.12.2021
Prognosen for den enkelte AML pasient avhenger av sykdommens karakteristika og pasientrelaterte faktorer som alder og komorbiditet (tabell 4.6 og vedlegg 1–4). I Norge benyttes risikostratifiseringen etter European Leukemia Net (ELN) der man vektlegger de genetiske avvik sammen med respons på første induksjonskur og grad av leukemisering. ELN har de tre risikonivåene god, intermediær og dårlig.
De genetiske avvikene inv(16)(p 13.1q22), t(16;16) (p 13.1;q22) og t(8;21)(q22;q22) er assosiert med en relativt god prognose; omlag 80 % av disse pasientene oppnår komplett remisjon og mindre enn 35 % får residiv etter kjemoterapi alene. Residivfrekvensen ved t(8;21) og leukocyttall over 20 x 109/L på diagnosetidspunktet er imidlertid høyere. Av de molekylær-genetiske avvikene er intern tandemduplikasjon (ITD) i FLT3 genet assosiert med dårligere prognose mens mutasjon i NPM1- og biallelisk mutasjon i CEBPA-genet har også uavhengig prognostisk utsagnskraft og er assosiert med god prognose (for ytterligere informasjon, se tabell 4.6). Allogen stamcelletransplantasjon i første remisjon er vanligvis ikke aktuell hos pasienter med «gunstige» kromosomavvik av de typene som er beskrevet tidligere, fordi prognosen ved kjemoterapi alene er relativt god (Cornelissen et al., 2012; Döhner et al., 2017).
For pasienter i intermediær og ugunstig prognosegruppe skal man som hovedregel alltid overveie allogen stamcelletransplantasjon for pasienter inntil 70–75 år. Disse pasientene bør søkes til vurdering ved Norsk gruppe for allogen stamcelletransplantasjon. Nytte ved allogen stamcelletransplantasjon må alltid veies opp mot risiko for terapirelatert mortalitet Dette gjelder spesielt eldre pasienter med komorbiditet eller påviste genetiske avvik som predikerer svært dårlig overlevelse.
Tidligere benyttet man også risikostratifisering etter samarbeidsgruppen. I tillegg til cytogenetiske og molekylærgenetiske endringer i leukemicellene er de kliniske parameterne leukocyttall og respons på første induksjonskur tatt med. I Norge er det enighet om at man ikke lenger benytter denne prognosemodellen som grunnlag med tanke på indikasjonsstillingen for allogen stamcelletransplantasjon i første remisjon.
Anbefaling:
- Man benytter European Leukemia Nets klassifisering av AML-assosierte genetiske avvik som grunnlag for den prognostiske vurderingen av pasienter med AML.
Tabell 4.6: ELN-klassifiseringen av molekylærgenetiske og cytogenetiske avvik med prognostisk utsagnskraft ved AML (APL er utelatt) (Döhner et al., 2017)
Genetisk gruppe | Avvik |
|---|---|
Gunstige | t(8;21)(q22;q22.1); RUNX1-RUNX1T1 inv(16)(p 13.1q22) eller t(16;16)(p 13.1;q22); CBFB-MYH11 Mutert NPM1 og ikke mutert FLT3-ITD eller med Flt3-ITDlavt allel ratio (definert som allel ratio <0,5*) Biallelisk mutert CEBPA |
Intermediær | Mutert NPM1 og mutert FLT3-ITDhøyt allel ratio (allel ratio ≥0,5*) Ikke-mutert NPM1 og ingen FLT3-ITD eller med FLT3-ITDlavt allel ratio* (ingen ugunstige cytogenetiske avvik) t(9;11)(p 21.3; q23.3); MLLT3-KMT2A (MLL); påvist t(9;11) er avgjørende dersom det samtidig påvises sjeldne, høy-risiko mutasjoner. Cytogenetiske avvik ikke klassifisert som gunstige eller ugunstige |
Ugunstige | t(6;9)(p 23;q34.1); DEK-NUP214 t(v; 11q23.3); KMT2A (MLL) rearrangert t(9;22)(q34.1;q11.2); BCR-ABL Inv(3)(q21.3q26.2) eller t(3;3)(q21.3;q26.2); GATA2,MECOM(EVI1) -5 eller del(5q); -7 -17/abnl(17p). Kompleks karyotype; defineres som ≥3 kromosomforandringer men ikke dersom man har t(8;21), inv(16), t(16;16) og t(9;11). Monosomal karyotype, defineres som en enkelt monosomi (ikke tap av X eller Y) sammen med minst en tilleggs-monosomi eller et strukturelt kromosomavvik (ikke core-binding factor AML). Ikke-mutert NPM1 med FLT3-ITDhøyt allel ratio. Mutert RUNX1 eller ASXL når man ikke samtidig har gunstig klassifisering. TP53 mutasjon (ofte samtidig kompleks eller monosomal karyotype). |
* FLT3-ITD allel ratio er i ELN definret som FLT3-ITD/FLT3-WT.
Generelle retningslinjer for intensiv AML terapi
Sist faglig oppdatert: 23.12.2021
Definering av behandlingsmål
Når diagnosen AML er stilt må det for alle pasienter med alder inntil omlag 80 år tas stilling til om pasienten er tjent med intensiv induksjonsbehandling. For å oppnå komplett remisjon må man gi cytostatika med så høy doseintensitet at behandlings-relaterte komplikasjoner kan bli livstruende selv ved optimal oppfølging. Terapi-relatert mortalitet er med optimal støtteterapi under 5 % ved god utvelgelse av yngre pasientene. Diagnostikk og behandling av pasienter som gis induksjonsbehandling, skal derfor alltid skje ved hematologisk seksjon på regionsykehus eller velutstyrt sentralsykehus med egen seksjon for blodsykdommer, gode blodbankressurser, forsvarlig vaktkompetanse og adekvat intensivkapasitet. Eldre pasienter har mer komorbiditet, tåler behandlingen dårligere og sykdommen responderer ofte dårligere på behandling enn hos de yngre.
Praktisk gjennomføring av intensiv AML behandling
Behandlingen fører til 2–4 ukers beinmargsaplasi med livstruende granulocytopeni og trombocytopeni. Pasientene trenger regelmessig erytrocyttransfusjoner og profylaktiske blodplatetransfusjoner for å holde platetallet over 10 x 109/L. De fleste vil trenge bredspektret intravenøs antibiotikabehandling, ofte også behandling mot mistenkt eller verifisert invasiv soppinfeksjon og total parenteral ernæring. Pasientene må derfor få lagt inn tunnelert sentralt venekateter helst før behandlingsstart.
Pasienter som ikke bør ha intensiv behandling
Hos enkelte pasienter står ikke risikoen forbundet med intensiv terapi i rimelig forhold til muligheten for å oppnå meningsfylt effekt av behandlingen. Dette kan være pasienter over 80 år eller yngre pasienter med andre alvorlige sykdommer. Man bør også være mer tilbakeholden hos eldre pasienter med AML der man av erfaring vet at man oftest ikke oppnår remisjon, for eksempel kompleks eller monosomal karyotype, AML sekundær til myelodysplastisk syndrom, kronisk myeloproliferativ neoplasi eller tidligere cytostatika/strålebehandling. Et alternativ for disse er ikke-intensiv behandling som sjelden gir komplett remisjon, men med et håp om en tidsbegrenset stabilisering og dermed akseptabel livskvalitet.
Fertilitet
Yngre pasienter kan fortsatt være fertile etter ordinær kjemoterapi. Stamcelletransplantasjon med myeloablativ forbehandling vil i de aller fleste tilfelle føre til infertilitet. For yngre pasienter og deres partnere bør man ta opp spørsmålet om nedfrysing av sæd, men dette må avveies i forhold til om det er medisinsk forsvarlig før behandling startes. Uttak, nedfrysing og bruk av nedfryst ovarialvev fra pasienter med nydiagnostisert AML eller etter oppnådd remisjon er en eksperimentell prosedyre som kan overveies hos yngre kvinner med bevart ovarialfunksjon og i komplett remisjon før myeloablativ allogen stamcelletransplantasjon.
Støttebehandling
Kvalmeprofylakse og behandling gis i tråd med vanlige retningslinjer (Roila et al., 2016). For å unngå tumorlyse-syndrom gis rikelig væske (minimum 2–3 liter i.v. hvert døgn), og allopurinol 300 mg daglig under induksjonsbehandlingen. Rasburicase er sjelden indisert ved AML, men kan overveies ved høyt blasttall i beinmarg og/eller blod.
Kontroll av remisjonsstatus
Blodtellinger og beinmargsutstryk med tanke på remisjonsstatus gjøres før start av hver konsolideringskur og før kondisjoneringen for både autolog og allogen stamcelletransplantasjon.
Intensiv induksjonsbehandling ved ikke-APL varianter av AML
Sist faglig oppdatert: 23.12.2021
Bakgrunn
Flere randomiserte studier har vist at daunorubicin gitt i tre dager med dagsdoser 90 mg/m2 kroppsoverflate gir høyere remisjonsfrekvens og bedre langtidsoverlevelse enn dagsdoser på 45 mg/m2 når daunorubicin kombineres med cytarabin infusjon (Luskin et al., 2016). Dette er vist i flere studier for pasienter opp til 65 år, mens effekten av økte daunorubicindoser er langt dårligere dokumentert for pasienter over 65 år, og en studie viste ingen sikker gevinst av økt daunorubicindose for denne aldersgruppen (Lowenberg et al., 2009). En randomisert studie har vist ingen forskjell mellom dagsdoser 60 mg/m2 og 90 mg/m2, men disse pasientene fikk alle også neste kur som inneholdt daunorubicin 50 mg/m2 i tre dager (Burnett et al., 2015b). En siste og mindre studie viste heller ingen forskjell mellom daunorubicin dagsdoser på 60 mg/m2 og 80 mg/m2, men også her inkluderte man antracyclin i konsolideringen (Vaezi et al., 2017).
En stor randomisert studie viste likeverdige resultater for daunorubicin i dagsdoser 90 mg/m2 og idarubicin 12 mg/m2 når begge medikamentene ble gitt i tre dager kombinert med cytarabin 200 mg/m2/døgn som infusjon i 7 dager (Lee et al., 2017). En annen studie gav ingen overbevisende holdepunkter for at økning av idarubicin 12 mg/m2/dag fra tre til fire dager gav bedre resultater i kombinasjon med cytarabin for pasienter i alderen 50–70 år (Pautas et al., 2010).
To større studier har undersøkt om forsinkelse av behandlingsstart påvirker prognosen. I den ene studien var mediantid fra diagnose til behandlingsstart 8 dager og man fant ingen effekt på prognosen (Bertoli et al., 2013), i den andre studien var mediantiden 4 dager fra diagnose til behandlingsstart og man fant en dårligere overlevelse for pasienter under 60 år ved forsinket behandlingsstart ut over 4 dager (Sekeres et al., 2009).
For behandling av pasienter med påvist FLT3-mutasjon vises til kapittel Behandlings hos pasienter med FLT3 positiv AML.
CPX-351 (Vyxeos liposomal) er en liposomal formulering av cytarabin og daunorubicin som er godkjent i behandling av terapirelatert myeloid neoplasi. I denne pasientgruppen har CPX-351 vist seg å gi en høyere andel pasienter som oppnår CR/CRi enn standard induksjonsbehandling. Grunnet akkumulasjon av liposomer i beinmarg gir CPX-351 noe høyere behandlingsrelatert mortalitet grunnet lengre aplasiperioder og større risiko for alvorlige bakterielle infeksjoner.
Etter vurdering av Vyxeos liposomal i Besluttningsforum den 26.01.20 ble det besluttet å ikke innføre Vyxeos liposomal til behandling av voksne personer med nylig diagnostisert, terapirelatert akutt myelogen leukemi (t-AML) eller AML med myelodysplasirelaterte forandringer (AML-MRC).
Gjennomføring av induksjonsbehandling
Det tas beinmargsaspirat på mellom 14–28 dager etter start av behandlingen, tidspunkt er avhengig av protokoll, se under. Dersom det er over 15 % blaster i beinmargsutstryket må man gi ny cytostatikaterapi. De fleste vil i dag regne at en kur som inneholder høy/intermediær dose cytarabin er beste alternativ for pasienter som ikke går i komplett remisjon etter første induksjonskur. I en slik situasjon vil man tilrå følgende alternativer:
- Pågående HOVON-protokoll: Her er det krav om beinmargsprøve mellom dag 17 og 28, så eventuelt ukentlig frem til man har bekreftet CR/CRi eller manglende respons. Dersom man på dag 17 eller på et seinere tidspunkt påviser økte blaster over 15 % starter man umiddelbart andre kur (se nedenfor).
- Mayer-protokollen: Behandlingen bygger på konsolidering med høydose cytarabin. Dersom man har økte blaster i beinmarg på dag 14–17 bør man gi enten andre kur som ved HOVON-regimet (se nedenfor) eller gi kur etter FLAG-Ida (se kapitlet om resistens/tilbakefall).
Dosejustering ved lever og nyresvikt
Cytarabin i doser som benyttes ved induksjonsregimene ovenfor trenger ingen justering ved lever eller nyresvikt. Det er ingen enighet om man skal justere antracyclindoser ved nyresvikt; noen store sentre gjør ikke dette mens andre justerer basert på kreatinin ved behandlingsstart:
Anthracyclin S-kreatinin (mmol/L) | % dose |
|---|---|
Normal kreatinin | 100 % dose |
<250 | 75 % dose |
>250 | 50 % dose |
Ved påvirket leverfunksjon bør man vurdere å dosejustere anthracyclin etter følgende retningslinjer basert på bilirubinverdi ved behandlingsstart:
S-Bilirubin (mmol/L) Anthracyclin | % dose |
|---|---|
Normal bilirubin | 100 % |
<50 | 75 % |
>50 | 50 % |
Det må understrekes at flere HOVON-studier har hatt som et krav at estimert GFR skal være over 60 for at pasientene skulle kunne inkluderes.
Behandling ved Akutt udifferensiert leukemi (AUL) og akutt leukemi med blandet fenotype (mixed phenotype acute leukemia, MPAL)
Det er ingen konsensus om hva som er optimal behandling for MPAL og AUL. Data fra observasjonelle studier viser høyere andel komplette remisjoner etter ALL behandling enn AML-behandling. For pasienter med påvist translokasjon MPAL med t(9;22)(q34.1;q11.2); BCR-ABL1 anbefales behandling med Hyper-CVAD med tillegg av tyrosinkinasehemmer. For pasienter uten t(9;22)(q34.1;q11.2); BCR-ABL1 anbefales induksjonsbehandling som beskrevet i kapittel om ALL.
(Wolach et al., 2015, 2017)
Inklusjon av et tredje medikament i induksjonsbehandlingen?
Midostaurin kan bli aktuelt å vurdere som et tredje medikament i induksjonsbehandlingen hos pasienter med Flt3-ITD og som ikke er aktuelle for stamcelletransplantasjon; medikamentet bør i så fall brukes som del av et helhetlig behandlingsopplegg (Stone et al., 2017).. Gemtuzumab kan også bli aktuelt spesielt for pasienter med CD33+ AML celler og lav eller intermediær risiko der man vet at stamcelletransplantasjon ikke vil være aktuelt i første remisjon (Hills et al., 2014).
Anbefaling:
Man tilrår følgende for intensiv induksjonsbehandling:
- Ved induksjonsbehandling hos pasienter opp til 65 år skal gis daunorubicin ≥60 mg/m2/d. Anbefalingen er daunorubicin 90 mg/m2 daglig i 3 dager eller idarubicin 12 mg/m2 daglig i 3 dager, begge kombinert med cytarabin 200 mg/m2 kroppsoverflate/døgn som kontinuerlig døgninfusjon i 7 døgn.
- Induksjonsbehandling hos pasienter i alderen 66–80 år etter individuell vurdering er daunorubicin 60 mg/m2/dag i 3 dager og cytarabin 200 mg/m2/dag i 7 døgn.
- Induksjonsbehandling bør startes så snart som mulig og senest 5 døgn etter at leukemi-diagnosen er stilt.
- Hos pasienter over 65 år med påvist CBF AML (inv((16)(p 13q22)/t(16;16)(p 13q22) eller t(8;21)(q22;q22), kan man vurdere tillegg av gemtuzumab ozogamicin.
- For pasienter med FLT3-ITD og FLT3-TKD mutasjon anbefales tillegg av midostuarin til induksjonsbehandlingen. Midostaurin 50 mg x2 gis da som da dag 8 til 21, se kapittel Behandlings hos pasienter med FLT3 positiv AML for konsolidereningsbehandling ved FLT3 mutert AML.
Konsoliderende AML terapi etter oppnådd komplett remisjon
Sist faglig oppdatert: 23.12.2021
Generelle retningslinjer. Man bør så langt mulig velge et helhetlig regime med veldokumentert effekt og toksisitet og ikke en individuell sammensetning av enkeltkurer. I praksis betyr det behandlinger en av følgende alterantiver:
- Hovon/SAKK protokoll
- Meyer protokoll
Begrunnede individuelle tilpasninger kan likevel bli nødvendige spesielt hos eldre pasienter samt pasienter med tilbakefall uten mulighet for kurasjon (se eget kapittel). Pasienter opp til 70–75 år skal vurderes med tanke på allogen stamcelletransplantasjon, og det er viktig at man velger et regime der de prognostiske parametrene er best mulig dokumentert og utprøvd i en pasientgruppe som er relevant for den enkelte pasient.
Bruk av MRD undersøkelser blir drøftet i et eget kapittel; prøver tas før induksjon og etter andre kur.
For pasienter med påvist FLT3-ITD mutasjon vises til eget avsnitt.
Konsolidering etter HOVON/SAKK
Ved HOVON/SAKK protokoll får alle pasienter etter induksjonsbehandlingen Induksjonskur nr. 2. På bakgrunn av cytogenetikk, MRD og komorbiditet besluttes det da om pasienten behandles med enten kombinasjonskur med mitoksantron og etoposid, autolog stamcelletranplantasjon eller allogen stamcelletransplantsjon.
Gruppen med god prognose
- HMAS-behandling, kondisjonering med busulfan og cyklofosfamid.
- Dersom ikke HMAS er mulig gir man kur nr 3; kombinasjonskur med mitoksantron 10 mg/m2/dag for dag 1–5 og etoposid 100 mg/m2/dag for dag 1–5.
Gruppene med intermediær
- Allogen stamcelletransplantasjon med familie- eller ubeslektet giver anbefales så sant mulig (se eget kapittel om allotransplantasjon).
- Dersom ikke allotransplantasjon gir man HMAS-behandling.
- Dersom heller ikke HMAS er mulig gir man som en tredje kur mitoksantron + etoposid slik som det er beskrevet ovenfor for pasienter med god prognose.
Gruppen med dårlig prognose
- Allogen stamcelletransplantasjon med familie- eller ubeslektet giver anbefales så sant mulig (se eget kapittel om allotransplantasjon).
- Dersom ikke allotransplantasjon gir man HMAS-behandling (se avsnitt ovenfor).
- Dersom heller ikke HMAS er mulig gir man som en tredje kur mitoksantron + etoposid slik som det er beskrevet ovenfor for pasienter med god prognose.
Pasienter som tilhører gruppen med relativt gunstig prognose er vanligvis ikke kandidater for allogen stamcelletransplantasjon i første remisjon. Pasienter som er kandidater for allotransplantasjon og har donor bør transplanteres så raskt som mulig etter oppnådd full remisjon (Döhner et al., 2017). Dette bør skje slik at pasienten slipper å få mer enn 1–2 konsoliderende cytostatikakurer for å unngå at toksiske effekter bygger på seg (Döhner et al., 2017). Til pasienter som i god prognosegruppe uten uttalt komorbiditet anbefales autolog stamcelletransplantsjon.
Første kur HOVON regime:
Induksjon 1: Idarubicin kombinert med cytarabin, se avsnitt om induksjonsterapi.
Andre kur HOVON regime er:
For personer under 60 år:
Ett av følgende to regimer, begge regimer er brukt i hovon studier, regime med dunorubicin er det som brukes i aktuelle Hovon studier:
- Konsolidering 1/Induksjon 2 (ad modum HOVON 102) gis med:
- Amsakrin 120 mg/m2 dag 4–6
- Cytarabin 1 g/m2 x 2 dag 1–6 (totalt 12 doser over 6 påfølgende dager)
eller:
- Konsolidering 1/Induksjon 2 (ad modum HOVON 132) gis alternativt som:
- Daunorubicin 60 mg/m2 dag 1 til 3
- Cytarabin 1 g/m2 x 2 dag 1–6 (totalt 12 doser over 6 påfølgende dager).
For personer 60 til 65 år gis:
- Cytarabin 1 g/m2 x 2 dag 1–6 (totalt 12 doser over 6 påfølgende dager).
Andre kur gis så snart som mulig hvis det er mer enn 15 % blaster i beinmargsutstryk på dag 17 etter start av induksjonsbehandling. Samme kur gis også ved blasttall under 15 %, men da gis kuren først etter hematopoietisk regenerasjon med platetall >100 x 109/L og nøytrofile >1.0 x 109/L.
Andre kur etterfølges av G-CSF som stamcellemobilisering før høsting og kryopreservering av stamcellegraft med unntak av pasienter som man allerede vet skal til allotransplantasjon i første remisjon.
I samsvar med HOVON 132 anbefales pasienter i god prognosegruppe høydose-behandling med autolog stamcellestøtte (HMAS)-behandling dersom dette lar seg gjennomføre (Vellenga et al., 2011). Pasienter i god prognosegruppe som anses ikke å tåle HMAS får konsoliderende behandling med kur 3 som inneholder mitoksantron og etoposid (se neste avsnitt).
Tredje kur 3: For pasienter ikke aktuellt for HMAS eller allogen stamcelletransplantasjon gis som følger:
- Mitoksantron 10 mg/m2 /dag for dag 1–5 og etoposid 100 mg/m2/dag for dag 1–5.
Konsolidering etter Mayer-protokollen med høydose cytarabin i gjentatte kurer. Alder til og med 60 år
Konsoliderende behandling med Meyer protokoll kan benyttes hos pasienter i god risikogruppe, eller til pasienter i andre risikogrupper som man mener ikke er kandidat for allogen stamcelletransplantsjon. Pasienter med funksjonsklasse 3 og 4 ved diagnose ble ikke inkludert i HOVON protokollene og for pasienter med alvorlig sykdomsmanifestjon og/ eller redusert organfunksjon med god prognose eller som ikke er kandidat for allogen stamcelletransplantasjon har Meyer protokollene vært et godt alternativ.
Dette regimet bygger på Mayer et al. sin studie der man benyttet induksjonskur basert på daunorubicin pluss cytarabin og med gjentatte kurer med høydose cytarabin (HDAC, 3 g/m2/dose) som konsolidering (Mayer et al., 1994). Basert på risikofaktorer for alvorlig nevrologisk toksisitet beskrevet i den kliniske studien gis denne behandlingen bare til pasienter i første remisjon som samtidig ikke er eldre enn 60 år, har kreatinin under 105 μmol/L og alkalisk fosfatase under 3 ganger øvre normalgrense:
Høydosert cytarabin (HDAC) 3 g/m4 x 2 gitt i.v. over 3 timer dag 1, dag 3 og dag 5, i gjentatte kurer med omlag 4 ukers mellomrom, inntil 4 kurer totalt (Mayer et al., 1994; Tangen et al., 2008)
Ved nyresvikt gjøres dosereduksjon av cytarabin til 1–1,5 g/m2. Dette regimet gir en 4 års residivfri overlevelse på 40–45 %; behandlingen er best dokumentert hos pasientene med gunstig og til dels intermediær risikogruppe (se tabell 4.6). Det må gis profylakse mot keratit med dexametason øyendråper under og to dager etter avsluttet behandlingen. Når man følger retningslinjene er cerebellar toksisitet sjelden. Imidlertid vil cerebellar toksisitet ofte være irreversibel og kuren må ved tegn på nevrotoksisitet avbrytes umiddelbart. Før hver kur gjøres blodtellinger, samt blod- og beinmargsutstryk for å sikre fortsatt remisjon.
Det er ikke et absolutt krav å gjennomføre 4 kurer; antall kurer og doseintensitet vurderes ut fra toksisitet etter tidligere kurer. Noen grupper hevder at 1–2 kurer med HDAC er tilstrekkelig. Andre mener at cytarabin doser på 2–3 g/m2 gir for høy toksisitet og at en intermediær dose cytarabin (IDAC) på 1 g/m2 er tilstrekkelig, men de studiene som ligger til grunn for det siste synspunktet benyttet cytarabin kombinert med andre cytostatika og ikke monoterapi. De norske erfaringene med HDAC regimet er gode (Tangen et al., 2008) og det er derfor rimelig fortsatt å anbefale dette.
Alder over 60–65 år – konsolidering med en kur intermediær cytarabin
Verdien av konsoliderende behandling hos denne pasientgruppen er omdiskutert. Gevinsten er i beste fall marginal. Hver enkelt eldre pasient må vurderes individuelt også under konsoliderende behandling som må avpasses etter pasientens respons og etter den toksisitet man har sett under induksjonsbehandlingen, og eventuelle toksiske skader som opptrer senere. Man bør unngå flere kurer ved alvorlig toksisitet.
Med utgangspunkt i studien HOVON-SAKK-AML 103 (HOVON103) for pasienter over 65 år kan man benytte studiens standard-arm for konsolidering med kun en enkelt kur med intermediær dose cytarabin (Lowenberg et al., 2009):
Cytarabin 1 g/m2 som infusjon over 6 timer, 2 ganger daglig i 6 påfølgende dager
(dag 1–6, totalt 12 doser.
Denne konsolideringskuren gis på bakgrunn av respons-evaluering på dag 17 etter start av induksjonskur. Ved tegn til persisterende sykdom (>15 % blaster) på dette tidspunktet gis IDAC-kuren umiddelbart. Beinmargsundersøkelse gjentas ukentlig og ved fortsatt lave blast-tall venter man med denne IDAC-kuren inntil man har oppnådd regenerasjon av perifere blodverdier. Denne IDAC-kuren må gis seinest 8 uker etter start av induksjonskuren.
Alder over 65–70 år – konsolidering med inntil 7 lavdose-kurer
Man benytter et regime med inntil 7 kurer med mer lavdosert kjemoterapi; denne behandlingen har vist overlevelsesgevinst i en randomisert undersøkelse sammenlignet med mer intensiv behandling (Gardin et al., 2007).
Behandlingen må imidlertid avpasses etter respons og toksisitet.
Behandlingen består av inntil 7 kurer med daunorubicin og cytarabin gitt hver 4. uke:
Dag 1: Daunorubicin 45 mg/m2 intravenøst
Dag 1–5: Cytarabin 60 mg/m2 som subkutan 12 timers infusjon på pumpe
Kuren kan gis poliklinisk. Antibiotika, transfusjoner og sykehusinnleggelse kan bli nødvendig, men behovet er mindre enn ved vanlig aplasibehandling (Gardin et al., 2007).
For pasienter sam man mener ikke tåler intensiv kjemoterapi eller allogen stamcelletransplantasjon
For pasienter med intermediær eller dårlig risiko leukemi, der man etter induksjon mener at ytterligere konsolideringsbehandling med intensiv kjemoterapi eller allogen stamcelletransplantasjon ikke er aktuelt grunnet stor risiko for prosedyre-relatert død har vedlikeholdsbehandling med azacitidine vist økt leukemi-fri overlevelse. Hos disse pasientene kan man overveie månedlig azacitidine i ett år (evidensgrad C) (Huls et al., 2019).
Pasienter i første remisjon tilbys konsolidering i samsvar med ett av følgende anbefalte regimer:
- Allogen stamcelletransplantasjon kan vurderes hos pasienter opp til 70–75 år.
- Pasienter inntil 65 år kan behandles i samsvar med HOVON-SAKK protokoll som hos noen pasienter vil inkludere autolog stamcelletransplantasjon.
- Alternativt kan pasienter inntil 60 år behandles med høydose cytarabin 3 g/m2 to ganger daglig dag 1, dag 3 og dag 5 i gjentatte kurer med om lag 4 ukers mellomrom, inntil 4 kurer.
- Alternativt kan pasienter over 60–65 år tilbys et regime med enten (i) en konsolideringskur basert på cytarabin intermediær dose; eller (ii) gjentatte kurer med lavdosert daunorubicin og subkutan cytarabin.
Allogen stamcelletransplantasjon
Sist faglig oppdatert: 23.12.2021
Allogen stamcelletransplantasjon i første remisjon er aktuelt for pasienter som har minst 35–40 % risiko for tilbakefall uten transplantasjon. Det betyr at pasienter med relativt gunstig prognose vanligvis ikke er kandidater for allotransplantasjon i første remisjon mens pasienter i intermediær og alvorlig prognosegruppe bør vurderes for allogen stamcelletransplantasjon. I de tilfellene det er aktuelt med transplantasjon bør dette planlegges snarest mulig etter indusert remisjon slik at pasienten slipper å få mer enn 1–2 konsoliderende cytostatikakurer for å unngå en stadig påbygging av behandlings-indusert toksisitet som kan øke risikoen ved transplantasjon og i verste fall umuliggjøre transplantasjon (Cornelissen et al., 2012; Döhner et al., 2017).
Allogen stamcelletransplantasjon har i utgangspunktet en risiko for transplantasjons-relatert mortalitet (TRM) på 15–20 % i første remisjon selv hos yngre pasienter. Det er gode holdepunkter for at man ved bruk av meget godt matchet ubeslektet giver (såkalt 10/10 match) oppnår like gode resultater som med bruk av HLA-identisk familiegiver. Til pasienter over 40 år og for yngre pasienter med høy komorbiditet brukes ofte doseredusert forbehandling, såkalte RIC (Reduced Intensity Conditioning) eller non-myeloablative transplantasjoner, som gir lavere risiko for mortalitet. Det er utarbeidet score-systemer (HCT-CI, EBMT og en egen RIC score) som vektlegger komorbiditet, alder og type donor for å predikere risikoen for TRM (se Vedlegg 1–3). Komorbiditetsvurderingen tar sikte på å avklare risiko for TRM, dvs. risikoen for å dø av komplikasjoner relatert til selve prosedyren. De to årsaksgruppene som bidrar til dette er (i) faktorer som er relatert til pasientens generelle helsetilstand og (ii) transplantasjons-relaterte faktorer inkludert sykdomsstadium og -varighet, alder og valg av donor. For å vurdere disse kan man bruke henholdsvis HCT-Comorbidity Index (HCT-CI) (Sorror et al., 2005) og EBMT score (Gratwohl et al., 2009). Allotransplantasjon benyttes når man vurderer den økte muligheten for residivfri overlevelse ved allotransplantasjon å være større enn risikoen for TRM, men samtidig må man også ta hensyn til muligheten for langtidskomplikasjoner, spesielt kronisk GVHD (Lee, 2017; Martin et al., 2015).
Pasienter med intermediær risiko for residiv, med lav komorbiditetsindeks og med lav risiko for transplantasjonsrelatert død kan også være transplantasjonskandidater. Pasienter som har egnet familiegiver og likevel ikke transplanteres i første remisjon, kan være kandidater for allogen stamcelletransplantasjon ved begynnende residiv og bør derfor følges nøye mens de er i første remisjon.
Behandlings hos pasienter med FLT3 positiv AML
Sist faglig oppdatert: 23.12.2021
Induksjonsbehandling og konsolideringsbehandling
For pasienter med påvist FLT3 mutasjon anbefales tillegg av midostuarin 50 mg x2 fra dag 8 til 21 ved induksjonsbehandling samt dag 8 til 21 ved påfølgende kondolideringskurer. For pasienter som behandles etter HOVONprotokoll betyr det både ved induksjon 1 og induksjon 2; for pasienter som behandles etter Meyer protokoll betyr det tillegg ved induksjonskur samt ved påfølgende konsoliderende kurer med høydose cytarabin.
Vedlikeholdsbehandling hos FLT3 positiv AML som ikke behandles med allogen stamcelletransplantsjon
For pasienter som ikke behandles med allogen stamcelletransplantasjon anbefales vedlikeholdsbehandling med midostaurin etter konsolidersingsbehandling. Midostaurin gis i total 12 28-dagers sykluser, uten pause mellom sykluser, dvs ca. 1 års behandling. Dette er anbefalt.
Vedlikeholdsbehandling hos FLT3 positiv AML som behandles med allogen stamcelletransplantasjon
FLT3-ITD positiv AML har 30 til 60 % risiko for tidlig tilbakefall etter allogen stamcelletransplantasjon. Vedlikeholdsbehandling reduserer risiko for tilbakefall og øker langtidsoverlevelse og flere studier har vist at vedlikeholdsbehandling med proteinkinase inhibitoren sorafenib reduserer risiko for tilbakefall og forbedrer overlevelse (Bazarbachi et al., 2020). Midostaurin ble ikke brukt som vedlikeholdsbehandling i Ratify studien etter allogen stamcelletransplantasjon. Hverken vedlikeholdsbehandling med midostaurin eller sorafenib er vurdert av Nye metoder. Så langt er datagrunnlaget best for sorafenib. I samsvar med EBMT retningslinjer anbefales Sorafenib som vedlikeholdsbehandling for FLT3 positiv AML etter allogen stamcelletransplantasjon. Behandlingen bør startes tidligst mulig etter engraftment. Ved alvorlig GVHD bør behandling stoppes.
Minimal restsykdom (MRD, Minimal Residual Disease)
Sist faglig oppdatert: 23.12.2021
Minimal restsykdom (Minimal residual disease, MRD) kan måles med væskestrømcytometrisk immunfenotyping (flow-MRD) eller molekylærgenetiske metoder/kvantitativ revers transkriptase PCR (genetisk MRD). Undersøkelsene kan gjøres både i beinmarg og perifert blod.
Det understrekes at man bare kan benytte standardisert metodikk utviklet spesielt for MRD analyser, og tolkningen av svarene må skje i nært samråd med laboratoriet som tilbyr diagnostikken og transplantasjonsavdelingen. Vektleggingen av resultatet bør individualiseres, med det kan være nyttig å ha MRD resultater som en del av beslutningsgrunnlaget når man skal avgjøre spørsmålet om allogen stamcelletransplantasjon hos pasienter med god og intermediær prognose (jfr. tabell 4.6). I studier benyttes MRD på følgende måter:
- Etter induksjonskurer for å indentifisere pasienter med stor risiko for residiv
- I forløpet etter induksjonsbehandling. Stigende verdier i målinger etter avsluttet terapi kan eventuelt være en tidlig indikasjon på senere residiv
- Etter transplantasjon for å vurdere om indikasjon for pre-emptive tiltak
Utenfor studier er det vanskelig å gi sterke anbefalinger for bruken av MRD undersøkelser så lenge man ikke har resultater fra flere prospektive undersøkelser. Unntaket er for pasienter med påvist NPM1 mutasjon. I to store kliniske studier brukte man MRD analyse basert på påvisning av mutert NPM1 etter andre cytostatikakur; i den ene undersøkelsen brukte man ikke påvisbar MRD i beinmarg etter 2 kurer mens den andre brukte minst 4-log reduksjon i perifert blod av transkripter sammenlignet med diagnosetidspunkt for å gruppere pasientene (Balsat et al., 2017; Ivey et al., 2016). Begge undersøkelsene viste at pasienter med restsykdom hadde en betydelig større risiko for tilbakefall enn pasienter uten restsykdom; pasienter uten restsykdom hadde tilbakefallsrisiko på 30–40 % og en samlet langtidsoverlevelse på 75–85 % mens tilsvarende tall for pasienter med restsykdom var for tilbakefall 60–80 % og langtidsoverlevelse 35–40 %
Generelle retningslinjer for genetisk MRD
Ved molekylærpatologisk MRD påvises tilstedeværelse eller fravær av spesifikke mutasjoner/fusjonsgener etter behandling for AML. Molekylærpatologisk MRD benyttes både til å vurdere indiksjon for allogen stamcelletransplantasjon og om det kan være aktuelt med pre-emptiv behandling etter allogen stamcelletransplantasjon. Molekylærpatologisk MRD er så langt kun aktuelt hos pasienter som har påvist ett av følgende genetiske avvik:
- NPM1-mutert AML (også aktuelt hos pasienter med samtidig påvist FLT-ITD)
- RUNX1-RUNX1T1 t(8;21)(q22;q22)
- CBFB-MYH11 inv(16)(p 13q22)/t(16;16)(p 13;q22)
AML med NPM1 mutasjon
Pasienter med normal karyotype med isolert påvist NPM1 mutasjon har tidligere vært klassifisert i god prognosegruppe. Nylige studier tyder imidlertid på at pasienter der man etter andre kur kan påvise NPM1 transkriptnivå ≥10-3 over har betydelig risiko for residiv, og har nytte av for allogen stamcelletransplantasjon i CR1. Det er viktig å pressisere at ≥10-3 regnes som MRD positiv, mens pasienter med påvisbar transkript under 10-3 regnes som MRD negative. Pasientene med påvisbar NPM1 transkript under 10-3 har økt risiko for tilbakefall, men ikke indikasjon for transplantasjon.
Tidspunkter og situasjoner der MRD analyse bør utføres:
- Etter induksjonsbehandling for å vurdere indikasjon for allogen stamcelletransplantasjon Prøven tas etter kur nr. 2, det vil si:
- Etter andre induksjonskur dersom man følger induksjonsbehandling etter HOVON med dobbel induksjon
- Etter 1 kur høydose cytarabin for pasienter som får konsolidering med høydose cytarabin (Mayer-regimet).
- Etter siste konsolidering med kjemoterapi eller autolog stamcelletransplantasjon for å fange opp tidlig residiv og indikasjon for allogen stamcelletransplantasjon.
MRD prøve tas hver tredje måned i to år. - Etter gjennomgått allogen stamcelletransplantasjon for å vurdere indikasjon for pre-emptiv behandling.
Prøve tas før allogen stamcelletransplantasjon og hver tredje måned i to år.
Praktisk gjennomføring:
Det sendes alltid både blod og benmarg
Blod: 6 ml rør EDTA
Benmarg: 3–5 ml benmarg tilsatt EDTA (Ved flowcytometri: heparin)
Ved prøvetakning bør sprøyten skylles med en liten mengde heparin. Straks etter prøvetakning overføres materialet til et EDTA-rør og blandes godt. Ved behov kan laboratoriet motta benmarg tilsatt heparin
Materiale må sendes straks etter prøvetakning. Prøven bør ankomme i god tid innen 24 timer. Prøver bør ikke tas fredag og dag før helligdag, ved unntak må prøve ankomme senest kl. 13:30.
RNA fra diagnosetidspunktet blir brukt som referanseverdi og må sendes til laboratoriet som utfører MRD-analysen.
Tolkning av resultater
Molekylærgenetisk MRD negativ prøve i Oslo (RNA basert analyse) regnes som:
I blod: Negativ uten påvisbart transkript
I benmarg: NPM1 transkrip Beregnet MRD <10-4 eller ikke påvisbar
Oppfølging
- Ved negativ prøve (benmarg under <10-4) videre kontroll. Hver tredje måned i to år.
- Ved suboptimal prøvekvalitet, anbefales kontroll etter 4 uker. Det er satt en grense på 10 000 kopier ABL1 for sikkert negativt resultat.
- Ved målt transkript 10-4 – 10-7: Kontroll etter tre måneder.
- Ved målt transkript ≥10-3: Rask kontroll etter 4 uker og forberedelse til ASCT eller annen behandling.
Pasienter med transkript over 10-3 i benmarg regnes som MRD positive. Så sant det ikke foreligger kontraindikasjon anbefales allogen stamcelletransplantsjon. Ved påvisbar transkript under dette nivået er pasienten NPM1 MRD negativ, men har økt risiko for residiv og bør følges som beskrevet over
Ved påvisbar transkript 10-4 – 10-7 etter kur nr. 2 er det usikkert hvilken konsoliderende behandling som er best. Disse pasientene har økt risiko for residiv, men har (ved dette tidspunktet) ikke indikasjon for allogen stamcelletransplantasjon. Hos disse pasientene anbefales videre kjemoterapi fremfor autolog stamcelletransplantasjon siden allogen stamcelletransplantasjon vil tolereres dårligere på et senere tidspunkt.
AML med Core-binding Factor mutasjon (CBF AML og RUNX1-RUNX1T1 AML)
Hos pasienter med CBF mutasjoner kan respons følges med MRD. Imidlertid er evidens mtp bruk opp mot vurdering av indiksjon for allogen stamcelletransplantasjon ikke avklart. Det er kjent at pasienter med CBF leukemier kan ha påvisbar restsykdom med MRD i mange år etter induksjonsbehandling uten å oppleve residiv; råd under er derfor å ansees som ikke absolutte, men førende.
Krav til respons etter behandling: >3 log reduksjon av RUNX1-RUNX1T1 eller CBFB-MYH11. Det vil si lavere enn 0,1 % av nivået ved diagnostisk prøve. Følgende kriterier har vært anbefalt for oppfølging av pasienter med CBF-leukemi:
- Ved MRD positivitet (<3 log reduksjon/ >0,1 % av nivået ved diagnostisk prøve): kontroll etter 4 uker.
- Ved stigning <1 log: ny kontroll etter 4 uker og vurdering av ASCT eller annen behandling.
- Ved stigning >1 log kan man vurdere salvage/ASCT
Væskestrømcytometrisk vurdering av MRD
Bruk av væskestrømcytometrisk ved diagnosetidspunktet kan påvise en såkalt leukemiassosiert fenotype hos ca. 90 % som kan følges videre med en sensitivitet på 10–3 eller bedre. MRD negativ prøve regnes hvis det ikke kan påvises leukemiceller ved sensitivitet på 10–3.
Væskestrømcytometrisk påvisning av MRD er undersøkt i prospektive studier, bla HOVON/SAKK AML 42A studien (HOVON 42A) der MRD ble vist å ha uavhengig prognostisk betydning (Terwijn et al., 2013). Data mtp gevinst av å inkludere flow-MRD i beslutningsalgoritme mtp allogen stamcelletransplantasjon foreligger ikke. For pasienter med intermediær risiko har to studier, HOVON132 og GIMEMA AML 1310, vist at pasienter med MRD negativ sykdom behandlet med HMAS har like god langtidsoverlevelse som MRD positive pasienter behandlet med allogen stamcelletransplantasjon. I begge disse studiene var det få pasienter i hver gruppe og studiene var ikke designet for å vurdere om flow MRD kan benyttes til å differensiere behandling hos pasienter med intermediær risiko AML. Så langt anbefaler ingen internasjonale retningslinjer klart at man skal bruke flow-MRD til å differensiere behandling for IR-AML. Siden flow-MRD er en uavhengig prognostisk markør for IR-MRD, at EBMT anbefaler flow-MRD innført og at man forventer det vil bli brukt i beslutning mtp behandlingsvalg ved IR-MRD anbefales man at denne analysen etableres.
Praktisk gjennomføring av Flow-MR
Ved diagnose: Benmargprøve må sendes til laboratorium ved diagnose og laboratoriet må varsles om at flow-MRD skal settes opp.
Etter kur nr. 1 sendes ny prøve for å se om immunfenotype har endret seg.
Etter kur nr. 2 og før allogen stamcelletransplantsjon tas prøve Det er prøve på disse to tidspunktene som ansees som prognosisk.
Spesielle situasjoner
Sist faglig oppdatert: 23.12.2021
AML i svangerskapet
AML i svangerskap skal behandles ved regionsykehus; pasientene krever en individuell vurdering og et tverrfaglig samarbeid mellom fødselslege, nyfødtmedisiner og spesialist i blodsykdommer med spesiell erfaring i AML behandling. Man må ta hensyn til om pasienten har førstegangsdiagnose eller residiv av AML. I første trimester er svangerskapsavbrudd ofte blitt anbefalt. Intensiv AML behandling i andre trimester har økt risiko for komplikasjoner, men det er beskrevet vellykket AML terapi i denne delen av svangerskapet. I siste del av svangerskapet blir det en individuell avveining av behov for leukemiterapi opp mot mulighet for trygg forløsning (Lavi et al., 2014; Paydas, 2016).
Isolert myeloid sarkom
Dette er en sjelden manifestasjon som kan forekomme både primært eller som isolert tilbakefall også etter allogen stamcelle-transplantasjon. Man bør tilstrebe diagnostikk og behandling i samsvar med de retningslinjer som gis for ordinær AML behandling med cytogenetikk, myeloid panel, immunfenotyping av knust biopsi. Dette innebærer ofte et tett sammareide mellom hematolog, labratorier og avdeling som utfører biopsi slik at biopsi hånderes korrekt med kortest mulig transporttid til labratorium
Ved påvist genetisk avvik behandles isolert myeloid sarkom etter tilsvarende ELN prognosestadium. Hos pasienter uten påvisbare genetiske avvik, tidligere MDS, dvs der man ikke kan tilordne til en risikogruppe etter ELN, er det konsensus om ikke å anbefale allogen stamcelletransplantasjon, men kjemoterapi som konsoliderende behandling i CR1. Pasientene bør vurderes med PET-CT mtp metabolsk respons. PET positiv lesjon bør biopsiers for å verifisere om det forerigger restyskdom eller ikke.
Effekten av strålebehandling er dårlig undersøkt; man kan etter individuell vurdering eventuelt gi stråleterapi mot området for primærtumor, spesielt om man har en restlesjon (Almond et al., 2017; Solh et al., 2016).
CNS profylakse og CNS sykdom
CNS sykdom er sjelden og rutinemessig spinalvæske-diagnostikk anbefales ikke. Risikofaktorer for CNS affeksjon er hyperleukocytose, visse cytogenetiske avvik som inv(16) og kromosom 11-avvik, monocytoid differensiering og lav alder. Ved klinisk mistanke om CNS affeksjon ved nyoppdaget AML gjøres cerebral CT eller MR og en diagnostisk spinalpunksjon etter start av behandling når pasienten ikke lengre har sirkulerende blaster. Det bør gjøres både cytologisk og væskestrømcytometrisk undersøkelse.
Det anbefales ikke rutinemessig CNS profylakse, men det kan være grunnlag for individuell vurdering hos enkeltpasienter med risikofaktorer dersom disse ikke konsolideres med høy eller intermediær dose cytarabin. Ved påvist CNS sykdom anbefaler man følgende (Dohner et al., 2010):
- Intratekal cytarabin 40–50 mg gis ved lumbalpunksjon to til tre ganger i uken inntil normal spinalvæske i to påfølgende prøver ved både cytologisk og væskestrømcytometrisk undersøkelse. Etter normalisering av spinalvæsken gis ytterligere 3 injeksjoner med samme dose.
- Dersom manglende effekt etter 2 uker, vurderes overgang til trippel intratekal behandling med metotrexat 12 mg og dexamethason 4 mg i tillegg til cytarabin.
- Hvis behandlingsintensjonen er kurativ, kan man vurdere å avslutte med CNS-bestråling (24 Gy mot hjerne, 18 Gy mot medulla) dersom man ikke planlegger allostamcelletransplantasjon og kondisjonering med helkroppsbestråling.
Hyperleukocytose og leukostase
Hyperleukocytose blir vanligvis definert som AML blaster i blod over 50 eller 100 x 109/L (Röllig et al., 2015). Leukostase defineres som kliniske symptomer på grunn av hyperleukocytose, det forekommer vanligvis først ved leukocytter over 50–100 x 109/L men kan unntaksvis ses ved lavere verdier. Dersom man har pasienter med (i) markert redusert allmenntilstand, (ii) dyspne i hvile, (iii) nevrologiske forstyrrelser i form av synsreduksjon/lesevansker, konfusjon, delir, somnolens eller hjerneblødning, eller (iv) organsymptomer forenlig med vaskulær okklusjon (f.eks. priapisme, myokardinfarkt) og disse symptomene ikke har noen annen forklaring, må man regne med at dette skyldes leukostase. Hyperleukocytose behandles etter følgende retningslinjer (evidensgrad D):
- Pasienter som planlegges å få intensiv induksjonsbehandling skal starte med denne behandlingen samme dag etter at nødvendig diagnostikk er sikret.
- Pasienter som ikke skal ha intensiv induksjonsbehandling starter straks med lavdose cytarabin subkutant.
- Pasienter med sannsynlig symptomgivende leukostase slik det er beskrevet ovenfor får leukaferese så sant ingen kontraindikasjoner.
- Leukaferese skal ikke benyttes ved APL fordi det kan forverre koagulopati.
- Man er tilbakeholden med blodtransfusjon ved hyperleukocytose/leukostase.
Anbefalinger:
Anbefalinger ved disse spesielle situasjonene:
- Ved AML i svangerskapet skal behandling skje ved Regionsykehus.
- Ekstramedullær sykdom diagnostiseres og behandles i samsvar med generelle retningslinjer for AML behandling.
- CNS affeksjon: Man gir ikke rutinemessig profylakse, behandling skjer ved intratekal cytostatika; initialt cytarabin monoterapi og ved utilfredsstillende respons kombinasjonsbehandling.
- Hyperleukocytose: Intensiv induksjonsbehandling startes snarest mulig, leukaferese kan vurderes men kun dersom man har symptomgivende leukostase. Hos pasienter som ikke skal ha intensiv kjemoterapi benyttes subcutan cytarabin som blastreduserende behandling.
Behandling av primært refraktær og tilbakefall av AML
Sist faglig oppdatert: 23.12.2021
Primært refraktær sykdom defineres som ikke oppnådd komplett remisjon etter to intensive induksjonskurer. Eneste mulighet for langtidsoverlevelse hos disse pasientene er allogen stamcelletransplantasjon. Ut fra en individuell vurdering av den enkelte pasient må man da avgjøre om man skal forsøke videre å oppnå komplett remisjon, eller om man skal benytte transplantasjon etter FLAMSA-RIC regimet hos pasienter som ikke er i remisjon.
Komplett remisjon før transplantasjon gir antagelig best langtidsoverlevelse og det er i Norge enighet om at pasienter antagelig best tjent med ytterligere induksjonsforsøk enn FLAMSA-RIC, men at man hos noen pasienter med hypocellulær marg og stabilt lav andel blaster over tid kan være kandidat for FLAMSA-RIC. Proliferativ sykdom med leukocytose over 10, infiltrasjon i benmarg over 60 %, intramedullær sykdom og CNS sykdom er assosiert med dårlig effekt etter FLAMSA-RIC.
I praksis vil det i aldersgruppen under 65–70 år som oftest bli aktuelt å forsøke å indusere remisjon Det er ikke allmenn enighet om hvilket terapiregime som er best mtp å oppnå remisjon. Regime 1 til 3 sidestilt. Nye data viser at kombinasjonsbehandling azacitidine/venetoclax kan oppnå høy andel med CR/CRi hos pasienter med primær refraktær sykdom. Effekt og tosksistet av azacitidine/venetoclax sammenlignet med konvensjonell behandling er så langt ikke avklart (Archimbaud et al., 1995; de la Rubia et al., 2002; Döhner et al., 2017; Ramos et al., 2015; Thol et al., 2015; Wahlin et al., 1997):
Regime 1: 5 dagers MAE kur (M5A5E5)
Amsakrin 150 mg/m2 kroppsoverflate gitt daglig i 5 dager på dag 1–5
Cytarabin 200 mg/m2/døgn som kontinuerlig døgninfusjon, dag 1–5
Etoposid 110 mg/m2/dag, gitt daglig dag 1–5
Regime 2: MEC
Mitoxantron 8 mg/m2 gitt daglig, dag 1–5
Etoposid 100 mg/m2 gitt daglig, dag 1–5
Cytarabine 1000 mg/m2 gitt daglig, dag 1–5
Dexametason øyendråper daglig som keratitt-profylakse t.o.m. dag 5
Regime 3: FLAG-Ida
Fludarabin 30 mg/m2 daglig, dag 1–5
Cytarabin 2000 mg/m2 daglig, dag 1–5
G-CSF 300 µg/m2 daglig, dag 0–6
Idarubicin 12 mg/m2 daglig dag 1–3
Dexametason øyendråper daglig som keratitt-profylakse t.o.m. dag 5
Azacitidine/Venetcolax
Hos pasienter med primært refraktær og tilbakefall av AML oppnår ca 60% CR eller CRi med Azacitidine/Venetcolax. Denne kombinasjonen er enda ikke vurdert av Nye metoder for denne indikasjonen. Hos pasienter som ikke er i remisjon etter induksjon 2 anbefaler man å søke fagdirektør om bruk av Azacitidine/Venetcolax som bro til allogen stamcelletransplantsjon.
Azacitidine/Venetcolax har noe dårligere effekt hos pasienter med påvist FLT3-ITD mutsjon og hos pasienter med proliferativ sykdom.
Azacitidine/Venetcolax gis med sykluslengde på 28 dager. Hvis CR eller CRi ikke er oppnådd etter 2 sykluser er ytterligere behandling ikke hensiktsmessig. Azacitidine gis dag 1 til 7, mens venetcolax gis kontinuerlig. Venetcolax gir uttalt neutropeni. Siden behandingen gis i kurativ intensjon avbrytes ikke behandlingen så lenge det ikke er livstruende infeksjon. Der er anbefalt å gi Ventoclax i 28 dager for både syklus 1 og syklus 2. Tillegg av G-CSF kan benyttes (Jonas et al., 2019; Samra et al., 2020).
Det anbefales at man under behandlingen gir profylaktisk behandling mot invasiv soppinfeksjon med Posaconazol. Grunnet interaksjon med Posaconazol må dose av venetoclax reduseres. Posaconazol startes dag 5, samtidig reduseres venetoclax
Azacitidine 75 mg/m2 subkutant fra dag 1 til dag 7
Venetoclax gis kontinuerlig fra dag 1 til 28 trappes opp.
Dag 1: Venetoclax 100 mg
Dag 2: Venetoclax 200 mg
Dag 3 og 4: Venetoclax 400 mg
Dag 5 til 28 (samme dag oppstart Posaconazol 300 mg): Venetoclax 100 mg
For å forhindre tumorlyse anbefales tilleggsbehandling med Allopur 300 mg daglig og at pasienter drikker 1.5 til 2L per dag. Risiko for tumorlyse er lav (under 5 %).
Behandling av refraktær sykdom ved FLT3 mutasjon
Gilteritinib er en tyrosinkinasehemmer med spesifikt effekt mot FLT3-ITD og FLT3-TKD. I en studie der gilteritinib var randomisert mot kjemoterapi hos pasienter med refraktær AML var CR/CRi i gilteritinib armen 34 % mot 15.3 % i kjemoterapi armen og gilteritinib var også bedre tolerert enn kjemoterapi.
Gilteritinib anbefales fremfor kjemoterapi. Imidertid er det ikke gjort noen sammenligning mellom gilteritinib og behandling med venetoclax/azacytidin.
Hos pasienter med residiv av AML som hadde FLT3 mutasjon ved diagnosetidspunktet må man verifisere at mutasjonen er til stede ved residiv, da en god andel av pasientene taper mutasjonen ved residiv. FLT3 mutasjon kan også oppstå de-novo ved residiv.
Behandling av refraktær sykdom ved IDH1 eller IDH2 mutasjon
Ivosidenib ved IDH1 mutasjon og enasidenib ved IDH2 mutasjon har vist å kunne gi henholdsvis 40 % og 20 % CR/CRi hos pasienter med refraktær AML og dermed kunne benyttes som en bro til allogen stamcelletransplantasjon. Begge medikamentene er godkjent av FDA, og er til vurdering hos EMA. Enasidenib er under hurtig metodevurdering hos Nye metoder.
Residiv under pågående konsolideringsterapi
Pasienter som får residiv under pågående konsolideringsbehandling eller i løpet av få måneder etter at denne er avsluttet, har svært dårlig prognose. Man oppnår sjelden ny remisjon, og en eventuell remisjon blir kortvarig. Dersom man i samråd med pasienten vil forsøke å indusere remisjon er regimene M5A5E5, MEC eller FLAG-Ida aktuelle å bruke dersom de ikke er gitt tidligere. Palliativ behandling med transfusjoner, antibiotika ved infeksjoner, og lav-toksiske cytostatika-kombinasjoner med sikte på å bremse sykdommen er ofte det beste alternativet. Ved oppnådd remisjon er allogen stamcelletransplantasjon den eneste behandling som kan gi realistisk håp om helbredelse. Dersom man velger å forsøke å indusere remisjon, må man raskt starte prioritert søk etter ubeslektet giver hos pasienter som ikke har familiegiver.
Residiv etter avsluttet konsolideringsbehandling
Ved residiv som oppstår mer enn 9–12 måneder etter avsluttet behandling kan man prøve det opprinnelige induksjonsregimet med anthracyklin og cytarabin dersom dette er forsvarlig i forhold til maksimaldose av anthracyklin og risiko for kardiotoksisitet. Alternativet kan ved slik risiko være M5A5E5 (se ovenfor). Pasienter som får residiv i løpet av de første 9 måneder etter avsluttet behandling vil sjelden oppnå remisjon på det opprinnelige induksjonsregimet, og man kan da velge ett regimene som er nevnt under primært refraktær sykdom hvis de ikke er gitt tidligere. Allogen stamcelletransplantasjon er eneste realistiske mulighet for kurasjon (Breems et al., 2005; Döhner et al., 2017).
Residiv etter allogen stamcelletransplantasjon
Pasienter med residiv etter allogen stamcelletransplantasjon har en alvorlig prognose. Behandling i denne situasjonen er avhengig av tid fra transplantasjonen til residiv og om pasienten har manifest GVHD. For noen pasienter med remisjon minst 6–12 måneder etter transplantasjon og hvor det er fravær av GVHD kan man vurdere ny transplantasjon hvis man oppnår komplett hematologisk remisjon etter ny induksjonsbehandling (Reikvam et al., 2013). For pasienter med komorbiditet, kortere remisjonsvarighet etter transplantasjon eller som ikke er i remisjon kan donor-lymfocytt infusjoner (DLI) være et alternativ hvis pasienten ikke har GVHD. DLI kan enten gis alene, etter induksjonsbehandling eller eventuelt kombinert med azacitidine (Craddock et al., 2016; Schroeder et al., 2015). Man vil som regel også samtidig forsøke reduksjon/seponering av eventuell immunsuppresjon. En pågående klinisk studie (Heinrich-Heine University, 2011) baserer seg på erfaringene fra en større retrospektiv studie (Schroeder et al., 2015) og man gir i denne studien azacitidine enten som 100 mg/m2 i 5 dager eller 75 mg/m2 i 7 dager med 28 dagers intervall. Denne behandlingen kan kombineres med DLI og i studien gis dette direkte i etterkant av tredje, femte og syvende azacitidinkur. Studiene har vist at omlag 20 % av pasientene oppnår hematologisk remisjon, men 2-års overlevelse er kun 10–15 %. Kortere overlevelse synes assosiert med tidlig tilbakefall (<6 måneder fra transplantasjonen) og høy prosent AML-blaster i beinmargen. Når DLI gis sammen med azacitidin administreres DLI etter andre behandlingsperiode med azacitidin; DLI infusjonen gis da 48 timer etter siste dose azacitidin.
For pasienter som opplever residiv etter langvarig remisjon, men som ikke er aktuelle for retransplantasjon eller DLI, kan ny induksjonskur vurderes etter de samme retningslinjer som for andre residivpasienter (se ovenfor) Disse pasienten vil ikke kunne kureres og intensjonen vil være å forsøke å oppnå en ny langvarig remisjon.
Behandling av residiv hos pasienter over 65–70 år
Intensiv kjemoterapi vil her ofte ha liten effekt og ofte vil pasienten være best tjent med ikke-intensiv eller palliativ behandling som sikrer best mulig livskvalitet nærmest mulig eller i hjemmet. Unntak kan være pasienter som har vært i remisjon lenger enn ett år, som er i god allmenntilstand uten komorbiditet, og som selv sterkt ønsker forsøk på å indusere ny remisjon etter nøye og nøktern informasjon. Man bør da spesielt vurdere å bruke regimer uten anthracycliner (se ovenfor).
Anbefalinger:
Anbefalinger for behandling av primært refraktær sykdom og residiv av AML:
- Alternativer for behandling av residiv under pågående behandling eller innen
9–12 måneder etter avsluttet behandling:- Induksjonsbehandling med alternativt kombinasjonsregime av cytostatika.
- Alternativt allogen stamcelletransplantasjon uten at man har oppnådd komplett remisjon.
- Allogen stamcelletransplantasjon som konsolidering etter oppnådd remisjon.
- Palliativ behandling.
- Residiv mer enn 9–12 måneder etter avsluttet behandling
- Induksjonsbehandling med anthracyklin/cytarabin.
- Allogen SCT som konsolidering ved oppnådd remisjon.
- Alternativt vurdere allogen stamcelletransplantasjon uten oppnådd remisjon.
- Palliativ behandling.
Sykdomsstabiliserende og palliativ behandling av AML
Sist faglig oppdatert: 21.12.2023
AML-rettet terapi
Anbefaling - behandling av pasienter som ikke kan få intensiv terapi:
Alle pasienter skal ha behandling med transfusjoner og antibiotika (Se anbefaling i retningslinje for bruk av antibiotika i sykehus). Azacitidine eller cytarabin kan benyttes som sykdom-stabiliserende terapi; azacitidin bør foretrekkes hos pasienter med høyrisiko cytogenetikk og multilineær dysplasi. Decitabine er et alternativ for pasienter med høyrisiko karyotype eller TP53 mutasjon.
Praktisk og begrunnelse:
Ikke-intensiv behandling er ofte det beste alternativ til eldre pasienter og pasienter som forventes ikke å tåle intensiv behandling, dvs. har en uakseptabelt høy risiko for behandlingsrelatert mortalitet. Med denne type behandling er muligheten for å oppnå remisjon liten, men man kan oppnå en bedret overlevelse. Demetylerende behandling med Azacitidin er i flere studier vist å være minst likeverdige med beste alternative terapi (primært lavdose cytarabin, LDAC) hos eldre pasienter (Burnett et al., 2007; Dombret et al., 2015; Fenaux et al., 2009; Fenaux et al., 2010; Kantarjian et al., 2012b), spesielt pasienter med høyrisiko cytogenetikk eller multilineær dysplasi i henhold til WHO klassifikasjon har bedre overlevelse med denne type behandling enn med lavdose cytarabin (Dombret et al., 2015).
Azacitidine/Venetoklaks regnes som førstevalg til pasienter som ikke ansees å tåle intensiv behandling og er godkjent av Nye metoder.
Azacitidine gis dag 1 til 7, mens venetoklaks gis kontinuerlig. For å forhindre tumorlyse anbefales tilleggsbehandling med Allopur 300 mg daglig den første uken. Pasienter som kan drikke inntar1.5 til 2L per dag, ellers må det gis intrevenøs hydrering.. Risiko for tumorlyse er lav (under 5 %).
Dosering:
Azacitidine 75 mg/m2 subkutant fra dag 1 til dag 7
Dag 1: Venetoclax 100 mg
Dag 2: Venetoclax 200 mg
Dag 3 og 4: Venetoclax 400 mg
Dag 5 til 28 (samme dag oppstart Posaconazol 300 mg): Venetoklaks 100 mg
Oppfølging under behandling:
• Dag 15 til 17 tas benmargsprøve og ved hypocellulær/acellulær benmarg stoppes venetoclax. Ved fortsatt økt andel blaster kontinueres venetoxlax til dag 28 trappes opp på følgende måte:
• Azacitidine/Venetoklaks gis med sykluslengde på 28 dager. Hvis CR eller CRi ikke er oppnådd etter 2 sykluser er ytterligere behandling ikke hensiktsmessig.
• Syklus 2 utsettes til nøytropeni og trombocytopeni er grad 2 eller bedre
• Dersom grad 4 trombocytopeni eller nøytropeni varer 7 dager etter at venetoklaks er stoppet, skal venetoklaks i syklus 2 gis kun i 21 dager
• Dersom grad 4 nøytropeni eller trombocytopeni varer 7 dager i syklus 3 eller senere, skal venetoklaks i påfølgende syklus gis i 7 dager kortere behandlingslengde
• Hvis pasienten har oppnådd varig CR eller CRi i 12 måneder, bør seponering av venetoklaks vurderes
Sopp-profylakse med aktivitet mot aspergillus species anbefales til pasienter med vedvarende neutropeni. Det er interaksjon med soppmidler og venetoclaks reduseres ved samtidig bruk av posaconazol til 70 eller 100mg daglig. Soppprofylakse pauses under opptrapping og startes dag 5 samtidig med redusert dose venetoclax
Decitabine (20 mg/m2/dag, daglig i 10 påfølgende dager dag 1–10, sykluslengde 28 dager) viste i en studie å ha en stabiliserende effekt spesielt hos pasienter med høyrisiko karyotype og hos pasienter med TP53 mutasjon; denne behandlingen kan derfor være et alternativ for denne pasientgruppen (Welch et al., 2016).
Lavdose cytarabin gir inntil 20 % komplett remisjon, men forventet 1-års overlevelse ligger bare på omlag 25 % og 8 ukers overlevelse er 60 %. Behandlingen må regnes å være uten effekt hos pasienter med høyrisiko cytogenetikk. Behandlingen tolereres oftest bra og kan gis poliklinisk. Det kan være hensiktsmessig at pasientene utstyres med tunnelert sentralt venekateter pga. muligheten for alvorlig infeksjon i aplasifasen med behov for i.v. væske, antibiotika og transfusjon.
Subkutane injeksjoner med lavdosert cytarabin (LDAC), 20 mg x 2 sc. daglig i 10 dager, eventuelt gjentatt hver 4–6 uke (evidensgrad A) (Burnett et al., 2007).
Ved svikt av azacitidin kan noen pasienter ha nytte av decitabine; de publiserte erfaringene er imidlertid at de fleste pasienter får ingen respons, rapporterte responser i litteraturen er ofte kortvarige og tvilsomme, og alvorlige bivirkninger er relativt hyppige (Duong et al., 2015b).
Understøttende/palliativ behandling alene
En del av pasientene vil ut fra en helhetlig vurdering ikke være aktuelle for verken intensiv eller non-intensiv behandling. Forventet levetid vil være kort. Pasientene bør kunne tilbringe mest mulig tid hjemme, delvis følges av fastlege/hjemmebaserte tjenester, og få behandling poliklinisk med blodprodukter og antibiotika.
Kliniske studier har vist at lavdose cytarabin, hydroxyurea eller 6-merkaptopurin kombinert med valproat og ATRA kan gi en stabilisering av sykdom vurdert etter MDS kriteriene med stigning spesielt av trombocytter også hos pasienter med høyrisiko sykdom etter konvensjonelle kriterier. Behandlingen gir lite bivirkninger og kan følges poliklinisk, den kan derfor være et supplement til annen palliativ terapi (Fredly et al., 2013a; Fredly et al., 2013b).
Almenntilstanden blir gjerne gradvis dårligere og det er behandlende leges ansvar å avslutte den palliative behandling. Dette må imidlertid gjøres i samråd med annet involvert helsepersonell og med pasient og pårørende.
Klassifikasjon, diagnostikk og prognosevurdering ved akutt promyelocytleukemi (APL)
Sist faglig oppdatert: 23.12.2021
Introduksjon
Akutt promyelocyttleukemi (APL) skiller seg fra andre leukemier ved uttalt blødningstendens som ubehandlet innen kort tid fører til fatal lunge eller hjerneblødning. Tidlig behandling med all-trans-retin syre (ATRA) og/eller arsenikk (arsentrioxid, ATO) reverserer koagulopatien, og langtidsoverlevelsen er over 80 %. Rask identifisering og umiddelbar behandling er avgjørende for å redusere tidlig mortalitet og dermed sikre god langtidsoverlevelse. Lavt platetall og blødningstendens er IKKE kontraindikasjon mot benmargsaspirasjon.
Hos de aller fleste pasienter med APL påvises translokasjonen t(15;17)(q24.1;q21.2) som medfører fusjon av vitamin A reseptor genet (RARA) med promyelocytic-leukemia-protein genet (PML). I WHO 2016 klassifikasjonen er leukemi med denne translokasjonen definert som en egen identitet; APL med translokasjonen t(15;17)(q24.1;q21.2);PML-RARA. Hos noen pasienter vil man ikke kunne påvise translokasjonen, men kun påvise fusjonsgenet PML-RARA. RARA kan imidlertid ha andre translokasjonspartner enn PML, for eksempel t(11;17) PZLF-RARA, t(5;17) NPM1-RARA eller t(17;17) STAT5b-RARA. Disse leukemiene har ofte et lignende klinisk og cytomorfologisk bilde som ved APL med PML-RARA, men effekten av ATO/ATRA er avhengig av RARA’s translokasjonspartner og behandling for disse variantene vil ikke bli nærmere omtalt i dette kapittelet.
APL utgjør omlag 5 % av alle akutte leukemiene, forekomsten av APL er lik i alle aldersgrupper og 5–20 % av APL-tilfellene er sekundære til tidligere cellegift eller strålebehandling. Pasienter med APL beholder sin gode prognose uavhengig av alder og om tilstanden er sekundær.
Den umiddelbare diagnostiske tilnærming
Behandling med ATRA skal startes umiddelbart ved klinisk mistanke, denne behandlingen skal fortsette inntil svar på genetisk diagnostikk har avkreftet diagnosen (ikke påvist translokasjonen t(15;17)(q24.1;q21.2) eller PML-RARA fusjonsproteinet). Undersøkelse med tanke på DIC skal gjøres umiddelbart (INR, APTT, fibrinogen, D-dimer, antitrombin) i tillegg til vanlige blodtellinger og blodutstryk. Typiske funn er hud- og slimhinneblødninger, betydelig trombocytopeni, økt d-dimer, nedsatt antitrombin og lavt fibrinogen. Normale verdier og fravær av blødinger utelukker imidlertid ikke APL.
Morfologisk diagnostikk
Omlag 75 % av pasientene har hypergranulær APL, svarende til tidligere FAB M3. Rikelig med blaster og promyelocytter med Auerstaver i benmarg forekommer hos de fleste men ikke i alle pasientene. Auerstaver hos pasienter med hypergranulær APL er ofte større og mer tallrike enn ved andre former for akutt leukemi. Omlag 25 % av pasientene har imidlertid en hypogranulær variant av APL (svarende til tidligere FAB M3var). Denne siste varianten har ofte ikke leukocytose, den er ofte vanskelig eller umulig å skille fra andre former for akutt leukemi basert på morfologi alene, men kan enkelte ganger ha karakteriske bilobære eller hjerteformete kjerner og eventuelt små blå granula i cytoplasma.
Væskestrømcytometri. Ved væskestrømcytometri finner man ofte karakteristiske trekk i forward/sidescatter. Den typiske immunfenotypen for blaster/promyelocytter er CD33+, CD34-, CD11b-/svak, HLA-DR- og CD117svak/ negativ; ingen av disse funnene ved væskestrømcytometri er imidlertid spesifikke eller sensitive nok til å utelukke APL, Den kliniske mistanken skal derfor være avgjørende for om man starter behandling med ATRA i påvente av endelig diagnostisk avklaring.
Cytogenetisk og molekylærgenetisk diagnostikk
Diagnosen APL verifiseres ved å påvise den spesifikke translokasjonen eller fusjonsgenet ved konvensjonell karyotyping, FISH eller PCR baserte analyser. Hvis laboratorium gjøres oppmerksom på problemstillingen vil svar på enten FISH eller PCR analyser foreligger som regel innen 2 dager. Gullstandard er påvisning av PML-RARA med revers transkriptase PCR (RT-PCR) og dette skal alltid utføres ved diagnose. RT-PCR analysen ved de forskjellige laboratoriene er optimalisert for et spesifikk antikoagulans i prøvene. Laboratoriet ved universitetssykehuset i Nord Norge og Radiumhospitalet benytter seg av EDTA-glass, Haukeland Universitetssykehus av PAX rør.
Etter avsluttet behandling benyttes kvantitativ RT-PCR av PML-RARA i benmarg som MRD markør etter avsluttet behandling; dvs. det er ikke nødvendig å utføre denne analysen før etter tredje konsolideringskur ved lavrisikosykdom og 2 uker etter avsluttet konsolideringsbehandling ved høyrisikosykdom.
Tilleggsdiagnostikk
Både CNS affeksjon og ekstramedullær sykdom forekommer hos pasienter med APL uten at dette ser ut til å endre prognosen. Det er kontraindisert å utføre spinalpunksjon eller biopsi før behandling er startet og man er helt sikker på at pasienten ikke lenger har koagulopati.
Prognostisk vurdering av pasienter med APL
Tidligere risikovurdering var basert på trombocytt og leukocyttverdi ved diagnosetidspunktet. Ved behandling med ATO har ikke trombocyttverdien prognostisk verdi og man benytter nå to risikokategorier.
- Lavrisiko APL: Leukocyttall ved diagnose ≤10 x 109/L
- Høyrisiko APL: Leukocyttall ved diagnose LPK >10 x 109/L
Ved APL har ca. 30 % FLT3-ITD mutasjonen, og andre cytogenetiske avvik i tillegg til t(15;17)(q24.1;q21.2 forekommer hos omlag 20 %. Både FLT3-ITD og/eller andre cytogenetiske avvik påvirker prognosen i så liten grad at det er bred konsensus om at slike tilleggsfunn ikke skal endre behandlingsopplegget.
Anbefalinger:
Den umiddelbare håndtering av pasienter med mistenkt APL:
- Alle norske sykehus skal ha all-trans-retin syre (Vesanoid/ATRA) tabletter tilgjengelig for umiddelbar bruk.
- Hos pasienter med akutt leukemi og der man ut fra klinisk bilde, morfologiske funn eller væskestrømcytometrisk undersøkelse mistenker APL, skal man umiddelbart starte med ATRA 45 mg/m2 fordelt på to doser daglig. Pasientene skal snarest mulig overføres sykehus med tilstrekkelig kompetanse for endelig rask diagnostisk avklaring og eventuell start av APL behandling.
- Ved klinisk mistanke om APL kontinueres behandling med ATRA frem til mistanken er endelig avkreftet med genetiske analyser.
- Ved mistanke om APL gjøres genetisk utredning som for andre former for AML. Imidlertid må genetisk laboratorium varsles slik at svar på analyse av PML-RARA fusjonstranskript kan foreligge snarest mulig, det vil si innen et par dager.
Støttebehandling ved akutt promyelocyttleukemi
Sist faglig oppdatert: 23.12.2021
Koagulopati
Å erkjenne tilstanden basert på en klinisk mistanke er avgjørende. Man starter da behandling med ATRA basert på klinisk mistanke alene. ATRA sammen med tidlig og tilstrekkelig substitusjon av blod og plasmaprodukter er avgjørende for å forhindre livstruende blødninger og dermed redusere den tidlige mortaliteten.
Fram til det ikke lengre er tegn til koagulopati skal man gi fibrinogenkonsentrater for å holde fibrinogen over 1,5 g/dl og transfusjoner av trombocyttkonsentrater for å holde trombocyttverdien over 30–50 x 109/L. I den første del av behandlingsperioden må fibrinogen og trombocyttverdi kontrolleres 3 til 4 ganger daglig.
Så langt fins det ikke data som støtter rutinemessig bruk av cyclokapron eller heparin.
Sentralvenøst kateter
Hos pasienter som er klinisk stabile og der man har god perifer venøs tilgang er det anbefalt ikke å legge sentralvenøst kateter eller Hickmannkateter før koagulopatien er under kontroll. Dette gjelder spesielt pasienter med lavrisiko APL hvor intravenøs behandling med ATO kan administreres via perifer venflon ettersom infusjonstiden av ATO kun er 2 timer.
Anbefalinger:
Generelle behandlingsretningslinjer ved APL:
- Hos pasienter med nydiagnostisert APL skal man gi tranfusjoner av trombocytt- og fibrinogen konsentrat for å holde trombocytter over 30–50x109/l og fibrinogen over 1,5 g/l fram til koagulopatien ikke lengre er til stede.
- Pasienter som etter oppstart av ATRA og eller arsenikk-trioksid (ATO) utvikler tegn på differesieringssyndrom skal umiddelbart starte behandling med intravenøs dexamethason 10 mg morgen og kveld.
- Vi anbefaler generelt ikke profylaktisk behandling med dexamethason, men det kan overveies hos pasienter med nyresvikt og/ eller leukocytter over 10 x 109/l.
- Pasienter med APL klassifiseres enten som lavrisiko-sykdom ved LPK <10 x 109/l eller som høyrisikosykdom ved LPK på diagnosetidspunktet ≥10x109/l.
Behandling av pasienter med lavrisiko APL
Sist faglig oppdatert: 23.12.2021
To randomiserte studier har vist at behandling med arsenikk og ATRA gir bedre resultater enn ATRA kombinert med kjemoterapi (Burnett et al., 2015a; Lo-Coco et al., 2013). Behandling med ATRA og arsenikk gir både lavere terapirelatert mortalitet og morbiditet initialt og synes å gi høyere leukemifri overlevelse. ATRA og ATO er derfor førstevalg ved behandling av lavrisiko APL uavhengig av alder, og man bruker det samme behandlingsregime uavhengig av alder.
Induksjonsbehandling
Denne behandlingen bygger på en kombinasjon av peroral ATRA og intravenøs ATO.
Tabell 4.7: Induksjonsbehandling ved lavrisiko APL
Medikament | Dosering | Dag |
|---|---|---|
ATRA Peroralt fra og med dag 1 | 45 mg/m2/dag fordelt på to doser daglig | Fra og med dag 1 og kontinuerlig inntil komplett remisjon, men maksimalt 60 dager |
ATO intravenøst Dag 1–5: | 0,3 mg/kg/dag | Dag 1 til 5, deretter dosert som angitt nedenfor |
ATO intravenøst Fra og med dag 6:* | 0,25 mg/kg/dag | Doseres 2 ganger i uken med 3/4 dagers mellomrom inntil 60 dager etter behandlingsstart. I tråd med dette gis altså ATO |
Hvis leukocytter stiger over 10 x 109/L startes hydroxyurea; se avsnitt om hyperleukocytose.
* Behandling med ATO avsluttes når pasienten oppnår komplett remisjon.
Ved APL reduseres andelen blaster og promyelocytter i beinmarg betydelig saktere enn ved andre former for AML og det kan ta inntil 60 dager før man oppnår komplett remisjon. Responsevaluering med benmargsdiagnostikk er derfor anbefalt først på dag 28 etter behandlingsstart. Hvis andel blaster/promyelocytter ikke er under 5 % på dag 28 kontinueres induksjonsbehandlingen og man gjør deretter ukentlige beinmargsundersøkelser. Induksjonsbehandlingen kontinueres fram til det er oppnådd komplett remisjon, eller til dag 60. Manglende remisjon etter induksjonsbehandling er svært sjelden ved lavriskosykdom (mindre enn 1 %). Spesielt i forbindelse med første uker av induksjonsbehandlingen må man være oppmerksom på risikoen for differensieringssyndrom.
Ved lavrisiko-APL er det ikke indisert med diagnostisk spinalpunksjon med mindre det foreligger kliniske symptomer på CNS sykdom. Det er ikke indisert med RT-PCR analyse av PMR-RARA for minimal restsykdom før etter 3. konsolideringskur.
Konsolideringsbehandling
Konsolideringsbehandlingen starter etter at pasienten har oppnådd komplett remisjon. Konsolidering 1, 2 og 3 er like med en sykluslengde på 8 uker, det betyr at påfølgende syklus starter 57 dager etter start av den foregående. Konsolidering 4 er litt kortere (4 ukers varighet) og inneholder noe mindre ATRA enn de tre første. MRD status tas etter konsolidering 3 før konsolideringssyklus 4.
Tabell 4.8
KONSOLIDERINGSSYKLUS 1–3 | ||
Medikament | Dosering | Dag |
Peroralt |
|
|
ATRA peroralt | 45 mg/m2/dag fordelt på to doser daglig | Dag 1–14 og dag 29–42 |
Intravenøst |
|
|
ATO intravenøst dag 1–5 | 0,3 mg/kg/dag | Dag 1–5 |
ATO intravenøst etter dag 6; 2 ganger ukentlig i uke 2–4 | 0,25 mg/kg/dag | To ganger ukentlig med 3/4 dagers mellomrom, altså dag 8, dag 11, dag 15, dag 18, dag 22 og dag 25. |
KONSOLIDERINGSSYKLUS 4 | ||
Medikament | Dosering | Dag |
Peroralt |
|
|
ATRA | 45 mg/m2/dag fordelt på to doser daglig | Dag 1–14 |
Intravenøst |
|
|
ATO intravenøst dag 1–5 | 0,3 mg/kg/dag | Dag 1–5 |
ATO intravenøst etter dag 6; 2 ganger ukentlig i uke 2–4 | 0,25 mg/kg/dag | To ganger ukentlig med 3/4 dagers mellomrom, altså dag 8, dag 11, dag 15, dag 18, dag 22 og dag 25 |
Oppfølging etter behandling med ATO og ATRA
MRD evaluering med RT-PCR fra benmarg skal tas etter konsolideringskur 3. Pasienter som er MRD positive etter tredje konsolideringssyklus skal vurderes for allogen stamcelletransplantasjon. Pasienter som er MRD negative er ikke aktuelle for vedlikeholdsbehandling.
For pasienter med lavrisiko-APL er sannsynligheten for residiv svært lav. Nytten av MRD er omdiskutert.
Anbefalinger:
Behandling av pasienter med lavrisiko APL:
- Pasienter med lavrisiko-sykdom behandles uansett alder med ATRA og ATO.
- Hos pasienter med lavrisikosykdom er det ikke aktuelt med vedlikeholdsbehandling ut over 4 konsolideringskurer med ATRA og ATO. Det er kun anbefalt 1 MRD prøve som tas etter tredje konsolideringskur.
Behandling av pasienter med høyrisiko APL
Sist faglig oppdatert: 23.12.2021
For høyrisiko APL foreligger det så langt ingen studier som direkte sammenligner ATRA og ATO med kombinasjons av cytostatika. Apollo studien er en pågående randomisert studie der resultatene forventes først å foreligge etter 2022. Imidlertid viser flere observasjonelle studier at ATRA og ATO gir like god overlevelse som ATRA og høydose kjemoterapi. ELN sidestiller nå disse to behandlingsformene (Sanz et al., 2019). Grunnet toksisitet assosiert med ATRA og cytostatika anbefales nå ATRA og ATO med tillegg av 4 dose antracyclin
ATRA, ATO og idarubicin
Dette regimet stammer fra en enarmet australsk studie (Iland et al., 2012). Resultatene i denne studien er gode og sammenlignbare med kjemoterapi kombinert med ATRA/kjemoterapi. Behandlingen består av en induksjonskur, 2 påfølgende konsolideringskurer og en avsluttende vedlikeholdsbehandling.
Tabell 4.9: Behandling av høyrisiko APL basert på ATRA, ATO og idarubicin
Induksjonskur | ||||||||||
Medikament | Dosering | Dag | ||||||||
Peroralt |
|
| ||||||||
ATRA | 45 mg/m2/dag fordelt på to doser daglig | Dag 1–36 | ||||||||
Intravenøst |
|
| ||||||||
ATO | 0,15 mg/kg | Dag 9–36 | ||||||||
Idarubicin; i alt 4 dagsdoser dosert etter alder |
| Dag 2, dag 4, dag 6, dag 8 | ||||||||
| Konsolideringskur 1 | ||||||||||
| Medikament | Dosering | Dag | ||||||||
| Peroralt |
|
| ||||||||
| ATRA | 45 mg/m2/dag fordelt på to doser daglig | Dag 1–28 | ||||||||
| Intravenøst |
|
| ||||||||
| ATO | 0,15 mg/kg/dag | Dag 1–28 | ||||||||
| Konsolideringskur 2 | ||||||||||
| Medikament | Dosering | Dag | ||||||||
| Peroralt |
|
| ||||||||
| ATRA | 45 mg/m2/dag fordelt på to doser daglig | Dag 1–7, 15–21 og 29–35 | ||||||||
| Intravenøst |
|
| ||||||||
| ATO | 0,15 mg/kg/dag | Dag 1–5, 8–12, 15–19, 22–26 og 29–33. | ||||||||
Vedlikeholdsbehandling som skissert over (tabell 4.9).
ATRA, ATO og Gemtuzumab-ozogamicin
For pasienter med absolutt kontraindikasjon mot antracycliner kan man vurdere behandling med ATRA, ATO og gemtuzumab-ozogamicin etter AML-17 protokollen (Ravandi et al., 2009). Behandlingen er da lik som skissert for lavrisiko APL, men kun med tillegg av en infusjon av gemtuzumab-ozogamicin 6 mg/kg i løpet av de første 4 dagene av induksjonskuren.
Det er ikke anbefalt at man behandler høyrisiko APL etter behandling med ATRA og ATO som skissert for lavrisiko APL grunnet økt risiko for både differensieringssyndrom og hyperleukocytose. Det anbefales profylaktisk behandling med steroider som angitt over.
Anbefalinger:
Behandling av pasienter med høyrisiko APL:
- Pasienter med høyrisiko sykdom anbefales ATRA/ATO med tillegg av 4 doser idarubicin. Hos pasienter med kontraindikasjon mot antracyclin gis en dose gemtuzumab ozogamicin.
- Pasienter med høyrisiko sykdom skal følges hver 3. måned i 1 år etter avsluttet sykdom med MRD, så hver 6. måned i 2 år. MRD måles i benmarg.
Behandling av APL residiv
Sist faglig oppdatert: 23.12.2021
Pasienter som får påvist hematologisk eller molekylært residiv etter primærbehandlingen vil være aktuelle for re-induksjon. Pasienter under 70–75 år som på nytt blir MRD negative etter induksjon bør vurderes for autolog stamcelletransplantasjon mens pasienter som forblir MRD positive bør vurderes for allogen stamcelletransplantasjon (de Botton et al., 2005; Meloni et al., 1997; Sainty et al., 2000; Sanz et al., 2007). Som hovedregel bør alle pasienter med påvist residiv søkes til vurdering ved norsk gruppe for allogen stamcelletransplantasjon.
Valg av regime til reinduksjon er avhengig av hvilket regime de fikk som primærbehandling. Pasienter som fikk ATRA kombinert med cytostatika som første behandling bør få ATRA og ATO mens de som primært fikk behandling med ATRA og ATO bør få ATRA+kjemoterapi. Gemtuzumab i monoterapi gir en høy andel komplette remisjon ved APL. Pasienter som ikke er kandidat for ATRA/ATO eller ATRA+kjemoterapi-baserte regimer kan vurderes for gemtuzumab-basert behandling (Lo-Coco et al., 2004).
Pasienter som grunnet alder eller komorbiditet ikke er kandidat for allogen stamcelletransplantasjon vil også være kandidat for reinduksjon. Konsoliderende behandling må da diskuteres på individuell basis.
Anbefalinger:
Behandling av APL tilbakefall:
- Pasienter under 70–75 år som etter primærbehandlingen opplever residiv (molekylærgenetisk eller morfologisk) bør få reinduksjon og påfølgende stamcelletransplantasjon.
- Pasienter som etter induksjonsbehandlingen oppnår MRD negativ status er kandidat for autolog stamcelletransplantasjon, mens pasienter som er vedvarende MRD positive bør vurderes for allogen stamcelletransplantasjon så sant det ikke foreligger kontraindikasjon.
Komplikasjoner ved behandling med ATRA og ATO
Sist faglig oppdatert: 23.12.2021
Differensieringssyndrom (Retinoic acid syndrom, RAS)
Differensieringssyndrom sees ved induksjonsbehandling med ATRA og ATO. Det er kjennetegnet med væskeretensjon, hypotensjon, nyresvikt, serositt (pleura og/eller perikardvæske), lungeinfiltrater og lungesvikt. Moderat til alvorlig differensieringssyndrom sees hos opp til 30 % av pasientene, og ubehandlet kan alvorlig differensiringssyndrom bli livstruende. Det eksisterer ingen aksepterte diagnostiske kriterier eller graderingssystem, og tilstander er ofte ikke mulig å skille fra sepsis. Følgende symptomer kan forkomme og noen angir at man skal mistenke tilstanden dersom man har minst tre av symptomene feber, vektøkning, lungeinfiltrater på rtg. thorax, pleura/perikardvæske, hypotensjon eller nyresvikt.
Ved mistanke om differensieringssyndrom (uansett alvorlighetsgrad) skal man starte behandling med Dexamethason 10 mg intravenøst morgen og kveld (Tallman et al., 2000). Denne behandling kontinueres frem til symptomene er borte og pasienten har oppnådd komplett remisjon. Det er ikke nødvendig med profylakse ved påfølgende kurer.
Ved alvorlig differensieringssyndrom skal behandling med ATRA og ATO stanses inntil tilstanden bedres; behandling med ATRA og ATO kan da gjenopptas. Noen studier har benyttet profylaktisk behandling med steroider, men det er uenighet om dette er godt nok dokumenter til å innføres rutinemessig. Risiko for alvorlig differensieringssyndrom ser ut å være avhengig av leukocyttall ved diagnose og om det foreligger nyresvikt. Generelt anbefaler vi ikke profylaktisk behandling med steroider. Hos pasienter med høyrisiko APL (Lpk over 5–10 x 109/L) eller nyresvikt (kreatinin over 120 mmol/L) kan man imidlertid overveie profylaktisk behandling med dexamethason 10 mg intravenøst morgen og kveld (Kelaidi et al., 2009; Sanz et al., 2014).
Forlenget QT tid
Behandling med ATO kan gi forlenget QT tid og potensielt livstruende arytmier; QTc skal derfor monitoreres før hver infusjon. Hos pasienter med betydelig kardiogen komorbiditet bør man også overveie telemetri under behandling. Før infusjon av ATO skal nivå av kalium være over 4,0 mmol/L og magnesium over 0,7 mmol/L. Man bør unngå bruk av andre medikamenter som også kan forlenge QT tiden (for eksempel kinoloner eller visse antiemetika som ondansetron). Ved beregning av QTc skal man ikke benytte Bazzets formel. I den Italienske studien med ATRA og ATO ble følgene formel benyttet:
QTc=QT+0.154 x (1000 – RR)
ATO skal ikke startes hvis QTc er over 500 ms. Etter behandling med ATO kan QTc være forlenget i opptil en uke etter avsluttet infusjon.
Hyperleukocytose
Under behandling med ATRA og arsenikk er det ikke uvanlig at det tilkommer leukocytose. Fordi man er redd for at dette kan forverre koagulopatien eller utløse differensieringssyndrom er det anbefalt å starte med hydroxyurea etter følgende doseringstabell:
Lpk 10–50 x 109/L: 500 mg fire ganger daglig
Lpk over 50 x 109/L: 1000 mg fire ganger daglig
Alternativet er repeterte enkeltdoser cytarabin eller idarubicin. Ved APL er det ikke anbefalt å utføre leukaferese fordi det kan forverre koagulopatien.
Hepatotoksisitet
Stigning av ALAT, bilirubin og/eller ALP til mer enn 5 ganger øvre referanseområde observeres hyppig under induksjonsbehandling med ATO. Hepatotoksisitet er sjelden under konsolideringsbehandlingen. ATRA og ATO kan kontinueres frem til ALAT, bilirubin eller ALP stiger over 5 ganger øvre normalgrense. Når disse verdiene faller under 4 ganger øvre normalgrense kan man starte igjen med ATRA og ATO i halv dosering. Hvis man etter 4 dager (to doser ATO) ikke observerer ny stigning av leverprøvene, kan man igjen øke til full dose ATRA/ ATO.
Pseudotumor cerebri
Pseudotumor cerebri er en kjent bivirkning av ATRA. Tilstanden er karakterisert ved kvalme, svimmelhet, hodepine, synstap og funn av papilleødem og forhøyet intraspinalt trykk uten at man kan påvise annen åpenbar intracerebral årsak. Spinalpunksjon/intraspinal trykkmåling vil som regel være kontraindisert på grunn av økt blødningsrisiko. Diagnosen stilles klinisk etter at man har utelukket andre årsaker. Ved sterk mistanke om pseudotumor cerebri skal behandling med ATRA stoppes frem til symptomene har gått tilbake. Når dette har skjedd kan ATRA startes i halv dose, etter ytterligere en uke bør man forsøke å trappe opp til full dose.
Anbefalinger:
Generelle behandlingsretningslinjer ved APL:
- Hos pasienter med nydiagnostisert APL skal man gi tranfusjoner av trombocytt- og fibrinogen konsentrat for å holde trombocytter over 30–50x109/l og fibrinogen over 1,5 g/l fram til koagulopatien ikke lengre er til stede.
- Pasienter som etter oppstart av ATRA og eller arsenikk-trioksid (ATO) utvikler tegn på differesieringssyndrom skal umiddelbart starte behandling med intravenøs dexamethason 10 mg morgen og kveld.
- Vi anbefaler generelt ikke profylaktisk behandling med dexamethason, men det kan overveises hos pasienter med nyresvikt og/ eller leukocytter over 10 x 109/l.
Vedlegg 1. HCT-comorbidity index (Sorror et al., 2005)
Sist faglig oppdatert: 23.12.2021
Kriteriene som skal vektlegges er gitt i tabellen nedenfor; de enkelte tilstander som skal vurderes er definert og vektleggingen av den enkelte faktor er også angitt.
HCT-KOMORBIDITETSINDEKS (HCT-CI): POENGBEREGNING | ||
Komorbiditet | Definisjon | Poeng |
Hjerterytmeforstyrrelse | Atrieflimmer eller flutter SIC sinus syndromer Ventrikulær arytmi | 1 |
Annen hjertesykdom | Koronarsykdom med stenose i en eller flere arterier som krever behandling i form av medikamentell terapi, stenting eller bypass. Hjertesvikt, hjerteinfarkt Ejeksjonsfraksjon ≤0.50 | 1 |
Cerebrovaskulær sykdom | Kjent TIA Gjennomgått apopleksi | 1 |
Inflammatorisk tarmsykdom | Crohns sykdom eller ulcerøs kolitt | 1 |
Leveraffeksjon, mild | Kronisk hepatitt Bilirubinstigning inntil 1.5 ganger øvre normalgrense ASAT/ALAT forhøyet inntil 2.5 ganger øvre normalgrense | 1 |
Diabetes | Sykdom behandlet med insulin eller perorale antidiabetika (sykdom behandlet med diett alene skal ikke vektlegges) | 1 |
Overvekt | Voksne pasienter med BMI >35 | 1 |
Infeksjon | Dokumentert infeksjon eller feber av ukjent årsak som vil kreve terapi før, under eller etter start av kondisjonering | 1 |
Psykiatrisk sykdom | Angst eller depresjon som krever psykiatrisk vurdering eller terapi på transplantasjonstidspunktet | 1 |
Revmatologisk sykdom | SLE, rheumatoid artritt, polymyositt, MCTD, polymyalgia rheumatica | 2 |
Ulcus-sykdom | Behandlingstrengende sykdom | 2 |
Nyresvikt | Kreatinin >170 μmol/L, dialysekrevende nyresvikt eller før nyretransplantasjon. | 2 |
Moderat lunge-funksjonsnedsettelse | CO-diffusjonskapasitet eller FEV1 redusert til 66–80 %, dyspne ved lett aktivitet | 2 |
Hjerteklaffsykdom | Alle typer klaffefeil bortsett fra asymptomatisk mitralprolaps | 3 |
Tidligere solid tumor | Tidligere behandlet, unntatt hudkreft som ikke er melanom | 3 |
Alvorlig lunge-funksjonsnedsettelse | CO-diffusjonskapasitet eller FEV1 redusert til ≤65 %, dyspne i hvile, oksygen-krevende lungesykdom | 3 |
Leversykdom, moderat og alvorlig | Levercirrhose Bilirubinstigning >1,5 ganger øvre normalgrense ASAT/ALAT forhøyet >2,5 ganger øvre normalgrense | 3 |
Bruk av HCTI-CI i forbindelse med allotransplantasjon
Man legger sammen pasientenes samlede poengsum. Undersøkelser i samlemateriale som inkluderer pasienter med ulike diagnoser, tyder på at den transplantasjons-relaterte mortaliteten begynner å øke spesielt når pasientene får en score på 3 eller høyere. Tabellen nedenfor angir funn i et materiale av AML-pasienter transplantert i første komplette remisjon hovedsakelig med familiegiver (medianalder omlag 40 år):
HCTI-CI: Mortalitet uten relasjon til tilbakefall av grunnsykdom | |
Poeng | Transplantasjonsrelatert mortalitet etter 2 år |
0 | Omlag 10 % |
1–2 | Omlag 20 % |
≥3 | 35–40 % |
Dette er altså data for hvordan faktorer relatert til den generelle helsetilstanden vil påvirke risikoen for transplantasjonsretalert mortalitet. Tallene i tabellen ovenfor er relevante for en gruppe pasienter med relativt lav risiko for transplantasjonsrelatert død (relativt unge AML-pasienter i første remisjon transplantert med familiegiver). I tillegg til dette vektlegges risikofaktorer assosiert med transplantasjonen som vurderes ut fra den såkalte EBMT-score der vektlegging av alder antyder overlapping mellom disse vurderingene (Vedlegg 2). Forskjeller i risiko for transplantasjonsrelatert mortalitet som har sammenheng med den hematologiske grunnsykdommen er bare delvis definert inn i EBMT-score.
Bruk av HCTI-CI hos pasienter som får intensiv induksjonsbehandling
Tallene bygger på en undersøkelse av 177 pasienter over 60 år som ble gitt intensiv induksjonsbehandling; om lag halvparten av pasientene hadde en score tilsvarende ≥3. Tidlig død ble definert som død innen 4 uker etter start av kjemoterapi (Giles et al., 2007).
HCTI-CI: Tidlig død ved induksjonsbehandling av nydiagnostisert AML | |
Poeng | Forekomst av tidlig død |
0 | 3 % |
1–2 | 11 % |
≥3 | 29 % |
Vedlegg 2. EBMT score (Gratwohl et al., 2009)
Sist faglig oppdatert: 23.12.2021
Denne risikovurderingen supplerer HCT-CI klassifiseringen ved å vektlegge transplantasjons-assosierte faktorer Risikoen assosiert med EBMT-score representerer derfor delvis/hovedsakelig en tilleggsrisiko i forhold til HCT-CI vurderingen. Tabellene nedenfor viser (i) hvilke faktorer og hvordan de vektlegges, (ii) definisjon av sykdomsstadium for de vanligste sykdommene, og (iii) hvordan poengene gjenspeiler mortalitetsrisiko.
EBMT SCORE | |
Risikoparameter | Poeng |
Pasientens alder <20 år 20–40 år >40 år |
0 1 2 |
Donortype HLA-identisk søskengiver Annen donor |
0 1 |
Kjønnskombinasjon pasient-giver Kvinnelig giver til mannlig pasient Annen |
1 0 |
Tid fra diagnose til transplantasjon <12 mnd >12 mnd |
0 1 |
Sykdomsstadium Tidlig Intermediær Sein |
0 1 2 |
DEFINISJON AV SYKDOMSTADIUM I EBMT SCORE | |||
Sykdom | Tidlig | Intermediær | Sein |
Akutt leukemi | Første komplette remisjon | Andre komplette remisjon | Alle andre stadier |
Myelodysplastisk syndrom | Ubehandlet eller i første komplette remisjon | Andre remisjon eller partiell remisjon | Alle andre stadier |
KML | Første kroniske fase | Alle stadier unntatt første kroniske fase og blastkrise | Blastkrise |
Non-Hodgkins lymfom og myelomatose | Ubehandlet eller første komplette remisjon | Andre komplette remisjon, partiell remisjon, stabil sykdom | Alle andre stadier |
Basert på klassifiseringen er det mulig å oppnå maksimalt 7 poeng for høyrisiko-pasienter. Ut fra totaldata kan man anslå at transplantasjonsrelatert mortalitet basert på denne klassifiseringen blir:
EBMT RISIKO | |
Poeng | Forventet mortalitet (%) |
0 | 12 |
1 | 16 |
2 | 26 |
3 | 30 |
4 | 37 |
5 | 41 |
6, 7 | 45 |
Vedlegg 3. RIC-score (Versluis et al., 2015)
Sist faglig oppdatert: 23.12.2021
Dette systemet inkluderer parametre både fra HCTI-CI og EBMT score og med en differensiert vektlegging av parametrene. Systemet bygger på en undersøkelse av AML-pasienter transplantert med redusert kondisjonering i første remisjon, medianalder 58 år (variasjonsbredde 20–76 år).
Parameter | Definisjon | Poeng |
|---|---|---|
Infeksjon | Krever kontinuering av antibiotika etter transplantasjonen | 1 |
Lungesykdom | DLco og/eller FEV1≤65 % eller dyspne i hvile eller behov for oksygen | 1 |
Ubeslektet giver | Matched unrelated donor | 1 |
Donor CMV | Donor CMV IgG serologi positiv | 1 |
Leversykdom | Cirrhose, bilirubin >1.5 ganger øvre normalgrense, ASAT/ALAT >2.5 ganger øvre normalgrense | 1 |
Tid til transplantasjon | Tid fra diagnose til transplantasjon ≥6 måneder | 1 |
Ulcussykdom | Behandlingskrevende | 2 |
Inflammatorisk tarmsykdom | Chrons sykdom eller ulcerøs kolitt | 2 |
Hjerteklaffsykdom | Alle klaffesykdommer unntatt mitralprolaps | 2 |
Psykiatrisk sykdom | Depresjon eller angst som krever psykiatrisk konsultasjon eller behandling | 2 |
Alder | ≥60 år | 2 |
Arytmi | Atrieflimmer eller flutter, syk sinusknute-syndrom, ventrikkelarytmi | 2 |
Pasient CMV | Pasient CMV IgG serologi positiv | 2 |
Overvekt | Pasient med body mass index >35 kg/m2 | 2 |
Rheumatisk sykdom | SLE, rheumatoid artrit, polymyosit, MCTD, polymyalgia rheumatica | 2 |
Nyresykdom | S-kreatinin> 177 μmol/L, dialyse eller tidligere nyretransplantasjon | 2 |
For å estimere risiko for transplantasjonsrelatert mortalitet kan følgende tabell benyttes:
Poeng | Transplantasjonsrelatert mortalitet | Totaloverlevelse |
|---|---|---|
0–3 | 8 % | 69 % |
4–6 | 17 % | 60 % |
≥7 | 38 % | 43 % |
Vedlegg 4. Estimering av tidlig mortalitet hos pasienter over 70 år som får intensiv induksjonsbehandling for AML (Kantarjian et al., 2010)
Sist faglig oppdatert: 23.12.2021
Man undersøkte 446 pasienter med alder over 70 år som ble behandlet med intensiv cytarabin-basert kjemoterapi. Tidlig induksjons-relatert mortalitet ble definert som død innen 8 uker etter start av kjemoterapi. I multivariat-analyse identifiserte man 4 risikofaktorer:
- Alder over 80 år
- Kompleks karyotype (≥3 avvik)
- Redusert performance status (2–4 ECOG score)
- Nyresvikt (S-kreatinin 115 μmol/l)
Antall risikofaktorer | % pasienter i denne gruppen | % pasienter tidlig død |
|---|---|---|
0 | 28 % | 16 % |
1 | 40 % | 31 % |
2 | 23 % | 55 % |
≥3 | 9 % | 71 % |
Akutt lymfoblastisk leukemi (ALL) og lymfoblastlymfom hos voksne
Hva er nytt?
Sist faglig oppdatert: 21.12.2023
Blinatumomab.
Blinatumomab ble i 2021 godkjent på MRD-indikasjon av beslutningsforum:
”Blinatumomab (Blincyto) innføres som monoterapi til behandling av voksne med Philadelphiakromosomnegativ CD19-positiv B-prekursor ALL som er MRD-positiv ≥0,1% under følgende vilkår:
- sykdommen er i første fullstendige remisjon
- behandlingen gjennomføres i tråd med anbefalingene i Nasjonalt handlingsprogram for diagnostikk, behandling og oppfølging av maligne blodsykdommer, og kun èn kur blinatumomab skal benyttes etter konsolidering.”
En kur blinatumomab gitt over fire uker bør tilbys ved CD 19 positiv og Philadelphiakromosom (Ph) negativ sykdom og MRD >0.1% dag 29 eller dag 70-90 (etter konsolideringen/blokk B1). Kuren doseres i henhold til Felleskatalogen. Etter kuren følger en ukes pause før man gjenopptar behandlingen i den protokollen pasienten følger fra det tidspunktet man avbrøt for å gi blinatumomab (altså fra dag 92 for Eldreprotokollen og NOPHO IR-arm, og tredje blokk-kur i NOPHO HR-arm), se første figur i vedlegget. For dem med MRD >0.1% dag 70-90 er det indikasjon for allogen stamcelletransplantasjon, og blinatumomab brukes da som bro til transplantasjon til dem som vurderes som transplantable.
Registrering av pasientene i Web CRF.
Alle pasienter over 18 år med ALL bør registreres i Web CRF.
- Pasienter som er inkludert i ALLTogether eller NOPHO protokollen registreres i studienes databaser som før.
- Ph neg ALL 18-45 som er inkludert i ALLTogether: Registreres som før både i Castor og NOPHO databasen.
- Ph neg ALL 46-65: Registreres i NOPHO databasen.
- Pasienter over 65 år registreres i Eldreprotokollen som før.
- Alle andre pasienter registreres i egen norsk Web CRF.
Dette gjelder også pasienter med Burkitt leukemi over 18 år, Ph pos ALL 18-65 år og Ph neg ALL 18-65 år som ikke inkluderes i ALLTogether eller NOPHO.
Denne har samme brukerløsning som for Eldreprotokollen, med samme nettadresse, brukernavn/passord og kode. Pasientene må samtykke skriftlig til registrering
Tilgang kan fås ved å kontakte petter.quist.paulsen@stolav.no.
Diagnostisk utredning
Sist faglig oppdatert: 21.12.2023
Blod/beinmargsundersøkelse
Se kapittel Diagnostikk for praktisk håndtering av prøvetaking/prøverør/medium/forsendelse.
- Beinmargsaspirat for lysmikroskopisk undersøkelse og beinmargsbiopsi gjøres på alle pasienter.
- Cytogenetikk (G-båndsanalyse): Gjøre på alle.
- Molekylærgenetiske undersøkelser: Dersom pasienten behandles i henhold til behandlingsprotokollen NOPHO 2008 eller ALLTogether hører det med egne analyser, se protokollene. Lab må informeres om at pasienten har ALL og hvilken behandlingsprotokoll som skal brukes. Hvis pasienten skal behandles i henhold til ALLTogether må prøvene sendes til Radiumhospitalet. For dem som ikke inkluderes i disse protokollene bør det med FISH eller PCR undersøkes for t(9;22) / BCR-ABL1 og 11q23 / KMT2A (MLL)-rearrangement. Raskt svar på BCR-ABL (<1 uke) er ønskelig.
- Ved mistanke om Burkitt bør MYC-rearrangement undersøkes. Dette kan gjøres med FISH på tilsendt materiale for cytogenetikk, på utstryk eller på biopsi.
- Thiopurin methyl transferase (TPMT) genotyping: Det bør enten testes for de tre vanligste mutasjonene av TPMT genet, eller gjøres funksjonstesting. Flere laboratorier i Norge kan brukes (St Olavs Hospital, UNN, Rikshospitalet (felles rekvisisjon).
- NUDT15 genotyping på de to vanligste mutasjonene bør gjøres på pasienter med asiatisk bakgrunn (Rikshospitalet)
- Immunfenotyping/DNA-ploiditet: Immunfenotyping ved flowcytometri gjøre på alle. Ved inklusjon i NOPHO skal det også gjøre DNA-ploiditet (indeks) ved flowcytometri.
- Minimal restsykdomsmonitorering (MRD): Man bør sende prøve både til Enhet for molekylærpatologi ved Radiumhospitalet og lokal flowlab. I ALLTogether brukes både flow og PCR til MRD-undersøkelse (høyeste verdi brukes). For pasienter som ikke behandles etter ALLTogether bør man også sende MRD-prøver til begge steder i tilfelle en av undersøkelsene skulle svikte teknisk, men ved B-ALL er immunfenotypisk undersøkelse tilstrekkelig som markør på framtidig restsykdom og ved T-cellesykdom brukes fortrinnsvis PCR av T-cellereseptor. Om slike markører ikke lar seg fremstille, brukes MRD ved den andre undersøkelsen. Opplys på remissen hvilken protokoll pasienten er tenkt inkludert i. For å få best mulig MRD-markør og god kvalitet på videre MRD-målinger er det avgjørende at man sender første del av benmargsaspiratet (2 EDTA glass i ALLTogether), i praksis betyr dette nytt og eget innstikksted.
- Vevstyping av pasienten og søsken/foreldre gjøres når det er klart at allogen transplantasjon kan bli aktuelt. For pasienter som behandles i hht. A2G protokollen skal dette gjøres ved diagnosetidspunkt.
- Det bør om mulig lages cellesuspensjon av leukemiceller som kryopreserveres for eventuelle senere kompletterende undersøkelser (lokal biobank, husk samtykke).
- Ved dry tap: Send biopsier til patologi, flow, cytogenetikk på McCoy og molpat. Send også perifert blod til flow, cytogenetikk og molpat.
Lymfeknutebiopsi
Lymfeknutebiopsi utføres ved lymfeknutesvulst hvor det er mistanke om B- eller T-lymfoblastisk lymfom eller Burkitts lymfom.
- En del formalinfikseres og sendes lokal avd. for patologi.
- En del av lymfeknuten sendes ufiksert til lokal avd. for patologi for supplerende diagnostikk som immunfenotyping, FISH/PCR og nedfrysing. Vurder også å sende en del på McCoy til cytogenetikk.
Øvrig utredning
Øvrig undersøkelse følger anbefalinger for lymfom/leukemiutredning, inkludert stadieinndeling (staging) av lymfoblastisk lymfom eller Burkitt lymfom. Spesielt viktig er:
CT (evt MR) thorax /abdomen/bekken ved mistanke om lymfoblastisk lymfom eller Burkitt lymfom. 18F-FDG-PET-CT anbefales ved responsevaluering dag 79 av lymfoblast lymfom. Man bør ideelt sett ha PET også fra før behandlingsstart, men ikke hvis det utsetter behandlingsoppstart hos pasienter der det er viktig å komme raskt i gang med behandling. Det er ikke krav om CT/MR ved ALL, der vil oftest klinisk undersøkelse inkl us av testis og rtg thorax være tilstrekkelig.
- CT/MR thorax ved tumor i mediastinum på rtg thorax.
- MR cerebri ved klinisk mistanke om CNS-affeksjon eller ved blaster i spinalvæsken.
- UL testis ved mistanke om testisaffeksjon.
Ved første intraspinale profylaksebehandling: Spinalvæskeundersøkelse med celletelling. Cytospin for alle pasienter uavhengig av celletall. Flowcytometri av spinalvæske er en egen studie i A2G, der flow brukes for å bekrefte positiv cytospin.
- LD, lever/galle-prøver.
- Virusserologi: EBV, CMV, HIV, HBV, HCV.
- Blodprøver ved behandlingsstart med tanke på utvikling av tumor lyse-syndrom (urat, Ca++, Mg++, K+, fosfat, karbamid, kreatinin).
Fertilitet
Sist faglig oppdatert: 21.12.2023
Nedfrysing av sæd før behandlingsstart bør tilbys menn i relevant alder når dette praktisk lar seg gjennomføre og utsettelse av behandlingsstart 1–2 dager kan forsvares medisinsk sett. Hvis dette ikke kan forsvares kan man sædbanke etter noen få dagers induksjonsbehandling. Mange blir ikke infertile av ALL behandling og det er viktig å bruke sikker prevensjon pga. teratogene medikamenter.
Nedfrysing av laparoskopisk uthentet ovarialvev kan vurderes før autlolog (HMAS) og allogen stamcelletransplantasjon (SCT) i samråd med fertilitetsklinikk. Dette er sjelden aktuelt ved diagnosetidspunkt på grunn av fare for leukemicellekontaminering, samt at sjansen for infertilitet ved kjemoterapi alene er mindre enn 50%.
Anbefalt behandling
Sist faglig oppdatert: 21.12.2023
Protokollene er tilgjengelige på Norsk selskap for hematologi (legeforeningen.no)
Philadelphia kromosom negativ lymfoblastisk leukemi
16–45 år
ALLTogether studien (A2G studien): For mer informasjon kontakt nasjonalt ansvarlige Hilde Skuterud Wik (OUS, Rikshospitalet).
Pasienter ≥ 16 år får alle samme induksjonsbehandling i ALLTogether (A2G) (induksjon B. Ved Down er det en helt egen behandlingsalgoritme, se protokoll). Videre behandling har tre behandlingsarmer. Følgende kriterier gjelder:
Standard risk.
- Ikke detekterbar MRD ved TP1 (dag 29) med PCR og flow.
- Ikke T-ALL
- Ingen høy-risk genetikk:
- KMT2A
- Hypodiploiditet <40 kromosomer
- iAMP21
- t(17;19)
- Ingen ABL-fusjon (ABL1, ABL2, PDGFRB, CSF1R)
- Ikke CNS3 eller traumatisk lumbalpunksjon
- Ikke blaster i CSF d 15
- Ikke testis eller mediastinal rester ved TP1
Intermediær risk høy.
Pasienter som ikke faller inn under kriteriene for SR- eller HR-armene, eller der man har mangelfulle opplysninger om MRD/mutasjoner/CNS-affeksjon.
Høy risk.
Pasienter ≥ 16 år med HR-kriterier skal behandles med allogen SCT i første remisjon. Dette kan erstattes av CAR-T for pasienter ≤25 år hvis de kan inkluderes i CAR-T studie. Indikasjoner (ett kriterium tilstrekkelig):
- T(17;19)(q23;p13)/TCF3-HLF
- MRD TP1 (d 29) ³ 5%
- MRD TP2 (d 71) ³ 0,01% ved B ALL (uavhengig av initial risikogruppe)
- MRD TP2 (d 71) ³ 0,05% ved T ALL
- MRD TP2 (d 71) ³ 0,05% ved ABL-fusjon
- Mediastinal masse som ikke er redusert til <1/3 av initialt volum ved TP2
- Vedvarende testis-affeksjon ved TP2.
- KMT2A-mutasjon: Etter diskusjon i nordisk ALL-gruppe for voksne anbefales SCT i første remisjon hvis MRD ved TP1 eller TP2 er over 0,01%. Øvrige pasienter behandles i IR høy armen.
Merk at protokollen er endret flere ganger fordi induksjonen og konsolidering 1 var langt mer toksisk enn i NOPHO 2008. Pasienter utviklet grav neutropeni og det var høy risiko for tromboser, myopati (dexametason), nevropati (vinkristin), hepatitt (asparaginase) og sepsis. Det er uvisst hvordan endringene slår ut. Man kan vurdere å la pasientene være innlagt fram til regenerasjon av neutrofile, og de bør få gjær- og muggsopp-profylakse med echinocandin under induksjonen (azoler interagerer med dexametason og vinkristin via cyp3A4). Sopp-profylaksen kontinueres i konsolidering 1 fram til neutrofil regenerasjon (to dager etter VCR/Dex kan echinocandin evt erstattes med posakonazol). For pasienter >25 år skal asparaginase dag 4 sløyfes. Asparaginase i konsolidering 1 er flyttet til senere i protokollen.
For dem som ikke er inkluderbare i ALLTogether: NOPHO 2008 protokoll, med tillegg av rituximab ved CD 20 positiv ALL (>20% positivitet) og blinatumomab ved CD 19 positiv sykdom og MRD >0.1 % dag 29 eller 79. Pasientene skal registreres i norsk Web CRF.
46–65 år (eller < 46 år som ikke skal behandles i henhold til ALLTogether)
Doseredusert NOPHO 2008 protokoll.
Inndelingen i behandlingsarmer som i originalprotokollen:
T-ALL og B-ALL med hvite over 100: Høy risk induksjon (dexametason). Øvrige: Standard risk induksjon (prednisolon).
B-ALL uten KMT2A og hvite <100 ved diagnose:
MRD d 29 <0,1% uten høy risk genetikk og uten CNS sykdom: Standard risk arm.
MRD d 29 0,1%-4,9% eller <0,1% og høy risk genetikk/CNS sykdom: Intermediær risk arm.
MRD d 29 ≥5%: Høy risk arm med SCT.
MRD d 79 ≥0,1%: Høy risk arm med SCT.
T-ALL og B-ALL med hvite >100:
MRD d 29 og 79 <0,1%: Intermediær risk arm.
MRD d 29 0,1%-4,9% og d 79 <0,1%: Høy risk arm med kjemoterapi (Blokk-kurer).
MRD d 29 ≥5%: Høy risk arm med SCT.
MRD d 79/post B1 ≥0.1%: Høy risk arm med SCT.
Hvis dag 15 benmarg med ≥25% leukemiske blaster: SCT hvis MRD ≥5 % etter blokk A1 eller ≥0.1% etter blokk B.
Pasienter med CD 19 positiv B ALL som har MRD ≥0.1% dag 29 eller dag 79: En kur blinatumomab etter konsolidering 1/Blokk B1 på toppen av NOPHO 2008.
Pasienter med KMT2A (MLL) avvik skal ha høy risk induksjon og de bør transplanteres i KR1 ved MRD >0.01% dag 29 eller dag 79. Øvrige pasienter behandles etter høy risk armen i NOPHO med dosejusterte blokker (evidens grad C).
>65 år
Norsk eldreprotokoll.
De aller fleste vil ha nytte av cytostatikabehandling, også de aller eldste og sykeste. Individualisering med dosereduksjoner etter behov og klinisk skjønn er viktig.
I induksjonen bør man unngå asparaginase pga. høy toksisitet, og hos eldre bør man velge pegylert asparaginase, som vanligvis tåles godt når det gis i konsolideringen. Stopp vinkristin ved uttalt nevropati/ileus. Høydose metotrexat tåles dårlig, max 500 mg/m2 hos eldre. Ved høy komorbiditet eller mye komplikasjoner kan man etter oppnådd remisjon overveie direkte overgang til forenklet vedlikehold (POMP eller 6-merkaptopurin/metotrexat alene) uten konsolidering.
B-celle terapeutiske antistoff tolereres godt i denne aldersgruppen, og anti-CD20 behandling (rituximab) bør kombineres med kjemoterapi ved CD20 positiv sykdom (>20%) uansett regime. En kur blinatumomab gis etter konsolideringen ved CD 19 positiv og Ph negativ sykdom og MRD >0.1% dag 29 eller dag 79. Deretter en ukes pause før man gjenopptar protokollen på dag 92 med intensifisering.
Ved Ph pos ALL legger man til TKI med imatinib 400–600 mg daglig. Pga interaksjon med asparaginase utelates asparaginase ved Ph pos sykdom. Hos de aller eldste og mest komorbide kan man gi induksjon bare med imatinib, steroider og vinkristin, og deretter vedlikehold med imatinib, 6-mercaptopurin og metotrexat.
Husk samtykke og registrering i Web CRF. Kontakt petter.quist.paulsen@stolav.no for samtykkeerklæring/CRF tilgang.
Philadelphia kromosom positiv ALL/lymfoblastlymfom
18–50 år:
HyperCVAD kombinert med imatinib 600 mgx1 til MRD <0.1%, så allogen SCT i KR1 (evidens grad B). Husk registrering i Web CRF.
51–65 år:
HyperCVAD kombinert med Imatinib 600 mgx1. Ved MRD<0.01% innen tre måneder kan SCT unnlates så lenge man beholder responsen (evidens grad B). Etter 2 år med POMP vedlikehold fortsettes TKI varig. Ved molekylært eller hematologisk residiv; allogen stamcelletransplantasjon. Hyper CVAD anbefales doseredusert til pasienter 60-65 år, se kurdetaljer i vedlegg. Husk registrering i Web CRF.
>65 år:
Norsk eldreprotokoll uten asparaginase og med imatinib 400–600 mgx1. Sistnevnte fortsettes på ubestemt tid etter avsluttet kjemoterapi (2.5 år) (evidens grad C). Husk registrering i Web CRF.
Etter allogen SCT bør man enten gi Imatinib 400 mg/d i minst to år profylaktisk eller gi TKI preemptivt. Hvis man velger sistnevnte anbefales det at man måler BCR/ABL i blod hver 3.–4. uke og i marg hver 6.–8. uke det første året, og starter 2. eller 3. generasjons TKI ved positiv prøve. Evidens grad B (Giebel et al., 2016; Pfeifer et al., 2013; Saini et al., 2020).
Dasatinib bør velges framfor imatinib ved CNS-affeksjon (Porkka et al., 2008).
Lymfoblastlymfom
18–65 år
NOPHO 2008, IR arm (doseredusert ved alder 46–65). Evidens grad B.
Ved manglende respons dag 29 (det er ikke krav om remisjon dag 29, men det skal være respons) eller manglende KR dag 79 anbefales eskalering til NOPHO blokker (A1, B1 og evt C1, med responsevaluering etter B1) og deretter allogen stamcelletransplantasjon (SCT) hvis man oppnår remisjon. Hvis man ikke oppnår remisjon på tre blokk-kurer anbefales strålebehandling mot resttumor før SCT. Utover dette anbefales ikke bruk av stråling ved LBL.
>65 år
Norsk eldreprotokoll
Burkitt og Burkitt lik lymfom/moden B ALL L3.
GMALL B-ALL/NHL 2002 med Rituximab (evidensgrad B).
GMALL B-ALL/NHL 2002 med Rituximab er best dokumentert og gir høye kurasjonsrater, også hos eldre. Første kur er toksisk, særlig for eldre, med fare for behandlingsrelatert død. Ved Burkitt lymfom uten benmarg eller CNS affeksjon kan dosejustert R-EPOCH være et alternativ for de aller eldste pasientene (evidens grad B/C). Se nasjonalt handlingsprogram for maligne lymfomer. Ved HIV-assosiert sykdom er det vesentlig å gi effektiv antiviral behandling sammen med cytostatikabehandlingen.
Husk at pasientene med Moden B ALL/Burkitt leukemi skal registreres i Web CRF.
Ved CD 20 positiv sykdom
ved CD 20 positiv ALL/lymfoblastlymfom (>20% positivitet) bør åtte doser Rituximab 375 mg/m2 med fire ukers intervall legges oppå aktuell protokoll (evidens grad B). Gjelder for alle aldre og både ved Ph pos og Ph neg ALL unntatt A2G protokollen.
CNS leukemi - ved diagnose
Ved leukemiske blaster i spinalvæske ved diagnose (bedømt ved cytospin eller flowcytometri) gis intratekal trippelbehandling med metotrexat 12 mg, cytarabin 30 mg og prednisolon 16 mg. Dette gis en til to ganger per uke (minst fire dager mellom hver injekson) til spinalvæsken er blastfri, deretter ukentlig fire ganger. Deretter gjenopptas vanlig profylakse i hht til protokollen som pasienten følger.
For A2G studien gjelder andre retningslinjer, se protokoll. Systemisk behandling fortsettes etter protokoll.
Ved hjernenerveaffeksjon eller parenchym affeksjon overveies CNS-bestråling 24 Gy, med 2 Gy fraksjoner, når remisjon er oppnådd.
Ved Ph pos ALL har dasatinib god overgang til CNS og bør velges.
Se også NOPHO 2008 og A2G protokollen for behandlingsforslag ved CNS leukemi.
CNS leukemi - ved relaps
Ved leukemiske blaster i spinalvæsken og samtidig beinmargsrelaps:
Intratekal trippelbehandling med metotrexat 12 mg, cytarabin 30 mg og prednisolon 16 mg gis to ganger per uke til spinalvæsken er blastfri, deretter ukentlig fire ganger. Deretter gjenopptas vanlig profylakse i hht protokoll. Protokollvalg avhenger av alder på pasient og hva som er gitt tidligere. Protokollen bør inneholde høydose metotrexat og/eller høydose cytarabin. Blokk-kurer fra NOPHO 2008 kan velges ved alder under 66 år. For pasienter over 65 år gjøres individuell vurdering, alternativer er aldersjustert hyper CVAD, Norsk Eldreprotokoll eller palliasjon.
Når man har oppnådd remisjon både i beinmarg og CNS gjøres CNS bestråling (24 Gy) etterfulgt av allogen stamcelletransplantasjon. TBI bør inngå i kondisjoneringen (12 Gy) og CNS-boost kan gis slik at man oppnår 24 Gy mot CNS.
Ved isolert CNS-relaps:
Ved isolert CNS relaps vil det bare vært et tidsspørsmål før det også kommer beinmargsaffeksjon, slik at det også må gis systemisk behandling. Men antall systemiske kurer blir ofte noe færre enn ved samtidig beinmargsrelaps. Ellers blir behandlingen som nevnt over.
OBS: For mange og hyppige i.t. kurer kan gi irreversibel medullaskade med varig parese/paralyse.
Etter at CNS bestråling er gitt bør i.t. behandling unngås pga toksisitetsfare.
Residivbehandling
- Ny remisjon og allogen stamcelletransplantasjon er nødvendig for kurasjon.
- ABC-blokkene i høyrisikoarmen i NOPHO 2008 protokollen kan være et godt valg ved residiv til dem under 65 år (evidensgrad C)
- Blinatumomab gir 40 % remisjonsrater ved resistent/relapsert ALL og er et aktuelt valg som bro til allogen stamcelletransplantasjon ved CD19 positiv sykdom ved manglende respons på kjemoterapi. Ved manglende MRD respons etter blokkbehandling kan blinatumomab være effektivt for å få ned nivået ytterligere før SCT. Når blinatumomab gis ved resistent/relapsert sykdom kan man vurdere å gi intratekal profylakse med metotreksat dag 15 under blinatumomab (Brown et al., 2021).
- Ved resistens på blinatumomab kan man forsøke inotuzumab (Kantarjian et al., 2016). Det er økt risiko for SOS/VOD i transplantasjonsforløpet etter inotuzumab og man bør gi lavest mulig dose, la det gå mest mulig tid mellom inotuzumab og transplantasjonen, og velge kondisjonering som gir lav risiko for SOS/VOD (Jabbour et al., 2018).
Hos pasienter som ikke er kandidater for allogen stamcelletransplantasjon blir behandlingen palliativ. Ofte kan man holde sykdommen i sjakk relativt lenge ved å gi ALL-aktive medikamenter som vinkristin, steroider, asparaginase, metotrexat og purinethol. Aktuelle cytostatikaregimer som kan modifiseres er OPAL, HyperCVAD eller NOPHO 2008.
CAR-T behandling med tisagenlecleucel (gis ved OUS, Rikshospitalet): Er aktuelt ved CD19 pos sykdom og alder <25 år ved residiv etter allo SCT, 2.gangs residiv eller senere uten allo SCT eller primær refraktær sykdom.
Allogen stamcelletransplantasjon
Sist faglig oppdatert: 21.12.2023
Generelle indikasjoner for allogen stamcelle transplantasjon (egne kriterier gjelder for NOPHO 2008 og A2G):
- Persisterende leukemi >5 % i beinmarg dag 29 etter behandlingsstart.
- Dag 70–90 MRD>0,1%
- Vedvarende positivt eller stigende MRD signal > dag 70–90.
- Philadelphia kromosom positiv ALL i KR1 ved alder <50 år. Ved alder >50 år: Ved MRD>0.01% dag 80-100
- KMT2A translokert ALL med MRD >0,01% dag 29 eller dag 70-80.
- KR≥2.
Alle som skal transplanteres bør helst ha MRD<0,1% før kondisjoneringen. Som bro til transplantasjon kan man enten bruke NOPHO 2008 blokker eller en kur blinatumomab. Det anbefales kondisjonering med total kroppsbestråling (Peters et al., 2020). Ved alder 46–65 er det samme indikasjoner for SCT som ved alder 18–45. Men i denne aldersgruppen bør redusert kondisjonering velges.
Om ALL
Sist faglig oppdatert: 21.12.2023
Ved mistanke om akutt lymfatisk leukemi/lymfom bør universitetssykehus kontaktes umiddelbart med tanke på overføring til øyeblikkelig-hjelp utredning og behandlingsstart. Dette er sjeldne sykdommer og det anbefales at behandlingen styres fra Universitetssykehus.
Lymfekreftsykdom som utgår fra rasktvoksende, umodne lymfatiske celler har store likhetstrekk enten sykdommen kalles akutt lymfoblastisk leukemi (ALL) eller lymfoblastisk lymfom. Etter WHO 2016-klassifiseres tilstander med mer enn 25 % lymfoblaster i beinmarg som ALL. Hvis beinmargskriteriet er oppfylt, kalles tilstanden altså leukemi selv ved stor mediastinal tumor. Ved lymfoblastisk lymfom (LB) i biopsi fra tumor eller lymfeknute og mindre enn 25 % blaster i beinmarg kalles tilstanden lymfoblastisk lymfom.
ALL kan deles inn i tre hovedgrupper; Philadelphiakromosom (Ph) positive, Ph negative og Burkitt lymfom/leukemi. Sistnevnte utgår fra modne B-lymfocytter og trenger spesiell behandling. Behandlingen avgjøres av typen tumorceller, og ikke om sykdommen manifesterer seg som leukemi eller lymfom.
For behandlingsvalget er det helt avgjørende med en mest mulig fullstendig morfologisk, immunologisk og cytogenetisk/molekylærgenetisk karakterisering av tumorcellene.
Disse sykdommene er ofte meget rasktvoksende, særlig Burkitt lymfom/leukemi og T-lymfoblast lymfom. Rask utredning med behandlingsstart i løpet av 1–2 døgn kan være vesentlig for å redde pasienter med stor tumormasse, f.eks. i mediastinum.
Spesielt ved Burkitt lymfom/leukemi foreligger det høy risiko for utvikling av tumor lyse syndrom. Så snart diagnosen er klar bør pasienten hydreres (ca 3000 mL/ /døgn). Ved Burkitt lymfom/leukemi, samt andre typer ALL med stort tumorvolum eller økt kreatinin bør det gis rasburicase (3-6 mg i.v., kan gjentas etter noen dager ved fortsatt høy urat (McBride et al., 2013)). Øvrige pasienter gis allopurinol (300 mg x 2/døgn til dag 7).
For aldersgruppen 16–45 år med Ph neg ALL har NOPHO 2008 protokollen vist gode resultater med 70-75 % 5 års overlevelse, som er ca. 25 % bedre enn Hammersmith 82 protokollen for tilsvarende aldersgruppe (Jæger et al., 2017; Toft et al., 2018). Men for pre B-ALL har det vært mange sene residiv, og blokkbehandlingen (som i hovedsak ble brukt ved T-ALL) medførte høy toksisitet. ALLTogether protokollen har nå erstattet NOPHO 2008 for denne aldersgruppen. Her er det meste av blokk-behandlingen sløyfet, MRD kravene skjerpet, og nye medikamenter introdusert (imatinib og inotuzumab). En utfordring vil være at asparaginase er inne igjen i induksjonen.
For pasienter mellom 45 og 65 år har det vært ca. 25 % langtidsoverlevelse i Norge med Hammersmith 82, med høy behandlingsrelatert dødelighet pga asparaginase i induksjonen (Jæger et al., 2017; Patel et al., 2017). Behandlingen for Ph neg ALL i denne aldersgruppen ble for noen år siden endret til doseredusert NOPHO 2008. Det er enda få pasienter som er inkludert, men så langt har det vært få tilbakefall og ingen toksiske dødsfall. Doseringene er utarbeidet i nordisk gruppe for ALL, og godkjent i alle nordiske nasjonale grupper. Man må innhente samtykkeerklæring fra pasientene og de skal registreres i NOPHO 2008 databasen.
Pasienter over 65 år har også hatt høy risiko for behandlingsrelatert død. Ved bruk av dosereduserte pediatriske protokoller kan man oppnå lavere toksisitet og over 30 % langtidsoverlevelse (Gökbuget, 2013; Martell et al., 2013). Intensiteten på cytostatikabehandlingen bør vurderes individuelt. Siktemålet for denne aldersgruppen er oftest livsforlengelse uten for høy behandlingsrelatert toksisitet. Behandlingsopplegg skal individuelt tilpasses etter alder, allmenntilstand, komorbiditet og behandlingstoksisitet. Norsk eldreprotokoll anbefales som et utgangspunkt.
T-ALL utgjør ca 1/3 hos voksne. Det kreves full pediatrisk protokoll inneholdende alle tradisjonelle leukemimidler for å oppnå gode resultater. Fem års overlevelsen med NOPHO 2008 for aldersgruppen 18-45 år var 65% (Quist-Paulsen et al., 2020).Det er få eller ingen tilbakefall etter mer enn 2.5 år (Quist-Paulsen et al., 2020). Svært få kan kureres ved tilbakefall (Quist-Paulsen et al., 2020).
ETP (Early T Precursor T) ALL. Leukemicellene har her beholdt immunfenotypiske og genetiske trekk fra myeloid linje og/eller stamceller. Typisk er sene MRD-responser, men til tross for dette har man med moderne pediatriske protokoller oppnådd like god overlevelse som øvrig T ALL (Quist-Paulsen et al., 2020).
Pre B-ALL. Det er denne gruppen som først og fremst skiller barn og voksne. For dem med CD 19 positiv sykdom og sen behandlingsrespons (MRD >0.1% dag 29 og <0.1% dag 79) er sene tilbakefall vanlig hos voksne med bare cirka 50 % hendelsesfri overlevelse etter 5 år med NOPHO 2008 protokollen (upubliserte data fra publikasjonskohort 2). Derfor anbefales en kur med blinatumomab etter konsolidering 1/blokk B1 (Gökbuget et al., 2018). Blinatumomab kan også brukes som bro til transplantasjon for å oppnå MRD negativitet (Gökbuget et al., 2018; Martinelli et al., 2017).
I ALLTogether protokollen har man skjerpet MRD kravet dag 71 til 0,01% og lagt til inotuzumab (randomisering).
Det er videre viktig å holde trykk på leukocytter/neutrofile under vedlikeholdsbehandlingen med metotrexat og purinethol tabletter. Husk at man IKKE skal gi folat-tilskudd ved ALL.
KMT2A ALL. Utgjør 5-10% hos voksne (Toft et al., 2018). Oftest Pro B fenotype, høye hvite og høy kinetikk med rasktvoksende sykdom, og også tidlige og rasktvoksende residiv. Linje/antigenskift kan forekomme ved residiv. Residiv er nesten umulig å kurere. Fra NOPHO 2008 kan det se ut som det er god prognose med blokk-kurer (høy risk kjemo arm) uten SCT for barn, samt for voksne med tidlig og dyp MRD respons (dag 29).
Philadelphia kromosom positiv ALL. Disse kan ikke inkluderes i NOPHO eller A2G. Til forskjell fra Ph neg ALL er dette en mer kronisk sykdom. Ofte bør man bruke TKI på varig basis etter kjemoterapi og evt SCT initialt. Selv med TKI og SCT er det relativt høy forekomst av residiv med langtidsoverlevelse rundt 50% (Saini et al., 2020). Det anbefales et regime med imatinib kombinert med cytostatika etterfulgt av SCT i første remisjon hvis mulig (evidensgrad B (Chalandon et al., 2015)). Det er uklart hva som er beste cytostatika, men hyper CVAD kombinert med TKI er best dokumentert (Sasaki et al., 2016). Hvor mye cellegift som trengs før SCT er ikke avklart, viktigst er trolig å oppnå MRD negativitet. SCT kan unnlates hos pasienter over 50 år (og pasienter <50 år med høy komorbiditet) som oppnår MRD<0.01% innen tre måneder (evidensgrad B) (Chalandon et al., 2015; Short et al., 2016), men det en enda uvisst om det er noe sikkert platå på overlevelseskurvene uten allogen transplantasjon. Man må gjøre individuelle avveininger etter pasientens ønske, komorbiditet, alder og respons. TKI og asparaginase interagerer og gir høy toksisitet. Skal ikke gis samtidig i induksjonen (Patel et al., 2017), og i handlingsprogrammet har vi valgt å unngå denne kombinasjonen også senere i behandlingen.
Burkitt lymfom/leukemi. GMALL B-ALL/NHL 2002 med Rituximab representerer den best dokumenterte behandlingen hos voksne med 80 % 5-års overlevelse (90 % for de unge og 60 % ved alder>55). Behandlingsindusert død i forløpet av første kur er høy hos dem med Burkitt leukemi og alder>55 år, og man kan vurdere å doseredusere A1 blokk til disse pasientene (Hoelzer et al., 2014). For de aller eldste pasientene uten benmarg og CNS affeksjon kan dosejustert R-EPOCH være et alternativ, særlig ved HIV/AIDS og samtidig antiretroviral behandling (Dunleavy et al., 2013; Roschewski et al., 2020).
Residiv. Til pasienter under 25 år som får residiv etter allotransplantasjon anbefales CAR-T behandling med tisagenlecleucel/Kymriah til dem med CD 19 positiv sykdom (Maude, 2018). Behandlingen er godkjent av Nye Metoder og gjøres på avdeling for blodsykdommer, Rikshospitalet.
Lymfoblastisk lymfom (LBL). Pediatriske protokoller gir meget god prognose ved LBL, bedre enn ved ALL (Cortelazzo et al., 2017). Vi anbefaler intermediær risiko armen av NOPHO 2008 (evidens grad C). Man vil da oppnå lavere risiko for sterilitet og sekundær AML/MDS enn tidligere behandlingsopplegg med Hammersmith etterfulgt av HMAS.
Diagnostikk og klassifikasjon
Sist faglig oppdatert: 21.12.2023
Diagnosen ALL baserer seg på påvisning av >20% blaster ved lysmikroskopering av benmargsutstryk, og med følgende immunfenotypiske funn (Arber et al., 2016):
- B-ALL. Minst to av disse: CD 19, CD 22, CD79, CD 10. Fravær av immunglobulin på overflaten.
- Moden B ALL (Burkitt): Immunglobulin eller lette kjeder på overflaten. MYC translokasjon kreves for å stille Burkitt diagnosen. Ved fravær av MYC bør/kan man stille diagnosen Burkitt-lik ALL og gi Burkitt behandling hvis morfologi, immunfenotype og klinikk ellers er passende med Burkitt.
- T-ALL: CD 3 positivitet cytoplasmatisk eller på overflaten.
- ETP (Early T Precursor T) ALL: Fravær av CD1a og CD 8, og tilstedeværelse av minst en myeloid eller stamcellemarkør (CD34, CD117, HLADR, CD 13, CD33, CD11b, CD 65).
WHO 2008/16 klassifikasjonen (Arber et al., 2016) deler ALL inn slik:
B lymphoblastisk leukemi/lymfom ikke nærmere spesifisert
B lymfoblastisk leukemi/lymfom med t(9;22)(q34;q11.2); BCR-ABL 1
B lymfoblastisk leukemi/lymfom med t(v;11q23); KMT2A (MLL) rearrangert
B lymfoblastisk leukemi/lymfom med t(12;21)(p13;q22) TEL-AML1 (ETV6-RUNX1)
B lymfoblastisk leukemi/lymfom med hyperdiploiditet
B lymfoblastisk leukemi/lymfom med hypodiploiditet
B lymfoblastisk leukemi/lymfom med t(5;14)(q31;q32) IL3-IGH
B lymfoblastisk leukemi/lymfom med t(1;19)(q23;p13.3); TCF3-PBX1
B-lymfoblastisk leukemi/lymfom med iamp21
B-lymfoblastisk leukemi/lymfom, BCR-ABL1–lik
T lymfoblastisk leukemi/lymfom
Tidlig T-ALL (Early T-cell precursor lymphoblastic leukemia)
For akutt udifferensiert leukemi (AUL) og ”mixed phenotype leukemi” (MPAL): Se AML kapittelet.
Prognostisk viktige undergrupper
Sist faglig oppdatert: 21.12.2023
NOPHO ALL-2008 og ALLtogether protokollen benytter egne risikofaktorer.
I tillegg til BCR-ABL og KMT2A er MRD den viktigste faktor å vektlegge med hensyn på prognose og indikasjon for allogen stamcelletransplantasjon (Malard et al., 2020). Hypodiploid karyotype (<45 kromosomer ved cytogenetikk eller DNA indeks <0.85 ved flow cytometri) og komplekse cytogenetiske avvik (≥5) er også dårlige prognostiske markører (Malard et al., 2020).
Responsevaluering
Sist faglig oppdatert: 21.12.2023
Minimal restsykdom (Minimal residual disease – MRD) i beinmarg
Man bør forsøke å definere leukemispesifikke markører med immunfenotypisk og/eller molekylærgenetisk metode ved diagnosetidspunkt, og følge MRD-nivået under behandlingen. De viktigste målepunktene er etter induksjonen (dag 29) og etter konsolideringen (dag 70–90). I HR armen i NOPHO følger man MRD før hver blokk-kur. Hos eldre kan man tilpasse individuelt, men man vil vanligvis nøye seg med MRD dag 29, dag 79 og etter 6 måneder. Hvis det er aktuelt med allogen transplantasjon og pasienten har høy risiko for tilbakefall anbefales MRD hver tredje måned i 1–2 år.
I NOPHO 2008 vurderes MRD som negativ ved verdier mindre enn <0,1 % (10-3). I A2G protokollen er MRD kravet for negativitet meget strengt og spesifikt beskrevet i protokollen, i praksis i området rundt 10-5. Ved Ph positiv ALL settes grensen til 10–4.
MRD <0,1% både dag 29 og 70-90 gir god prognose (ca 80 % langtidsoverlevelse både for B- og T-ALL ved NOPHO 2008). MRD >0,1% etter konsolidering dag 70–90 eller senere er knyttet til dårlig prognose, og gir indikasjon for allogen stamcelletransplantasjon (Vora et al., 2014). Positiv MRD dag 29 og negativ dag 70–90 gir middels prognose for B-ALL og god prognose for T-ALL (Quist-Paulsen et al., 2020).
Økende MRD (1 log eller mer) gir svært høy risiko for tilbakefall.
Ved positiv prøve på MRD gjentas prøven så snart det er praktisk mulig dersom beslutning om stamcelletransplantasjon skal baseres på prøveresultatet.
Ved overgang fra tidligere negativ til positiv status som ikke skyldes variasjon i test-sensitivitet bør pasienten om mulig bringes tilbake i MRD negativ status før allogen SCT. Kontakt Avdeling for Blodsykdommer, OUS, Rikshospitalet for planlegging av videre behandling.
Bildeevaluering ved lymfoblastlymfom
Man bør evaluere respons med CT/MR etter induksjonen (dag 29) og etter konsolideringen (dag 79). Etter induksjonen bør man ha oppnådd god tumor reduksjon, men minst 2/3 vil ikke være i komplett remisjon (KR). Etter konsolideringen bør det foreligge KR/KRu.
PET kan være en viktig undersøkelsesmodalitet for å bestemme viabilitet i mediastinal resttumor etter konsolideringen (evidensgrad C). Biopsi bør tas ved usikkerhet.
Noen med lymfoblastymfom vil ha benmargsaffeksjon, og man kan da i tillegg til billeddiagnostikk bruke MRD i benmarg for responsevaluering.
Antiinfeksiøs profylakse
Sist faglig oppdatert: 21.12.2023
Valtrex 250 mgx2 og Bactrim 1 tblx1 gis rutinemessig til alle fra behandlingsstart og til 3 måneder etter avsluttet behandling. Flukonazol 100–200 mg x1 gis i perioder med neutropeni og/eller steroider tilsvarende døgndoser >10 mg Prednisolon (eks NOPHO 2008 induksjon). Høyere doser flukonazol og alle doser av posakonazol interagerer med vinkristin (og dexametason) og kan føre til uttalt nevropati. Posakonazol profylakse gis i neutropeniperiodene etter blokkene i HR armen i NOPHO 2008. Man bør ha lav terskel for CMV PCR måling ved feber hos pasienter i HR armen i NOPHO2008. Under A2G induksjon bør det gis muggsopp profylakse, og pasientene bør være innlagt til regenerasjon av neutrofile pga høy toksisitet. Man kan gi echinocandin iv under induksjon og evt skifte til posakonazol i konsolidering 1 (gis til neutrofil regenerasjon).
Nyttige lenker til anbefalinger i nasjonal faglig retningslinje for bruk av antibiotika i sykehus:
Asparaginase
Sist faglig oppdatert: 21.12.2023
Asparaginase har svært høy toksisitet når det gis i induksjon (hepatitt, sepsis, tromboser), men tåles relativt godt ellers i behandlingen.
Problemer:
- Immunsvikt (cellulær og humoral).
- Trombose (inkludert sentralvene). Når asparaginase gis i induksjonen kan opp til 30% få trombose. Ved ikke livstruende VTE kan asparaginase gjenopptas etter noen uker under dekke av antikoagulasjon. Det er ikke vist at tromboseprofylakse er nyttig (Burke et al., 2020).
- Hepatitt (kan bli uttalt og gi leversvikt). Ved asparaginase-indusert hepatitt under induksjon kan asparaginase gjenopptas senere i behandlingen hvis hepatitten ikke var livstruende og har gått tilbake.
- Pankreatitt (inkludert nekrotiserende). Asparaginase bør vanligvis ikke gjentas etter en pankreatitt-episode (Burke et al., 2020). Ca 50% vil få residiv av pankreatitt ved reeksponering og faren for langtidskomplikasjoner er høy (DM, kroniske smerter, pseudocyster).
- Allergiske reaksjoner (inkludert anafylaksi). Etter en allergisk reaksjon er det ingen effekt av medikamentet lenger og man bør skifte til annet produkt (Erwinia- eller erytro-asparaginase hvis reaksjon på E-Coli asparaginase). Stille inaktivering kan forekomme og man bør følge asparaginaseaktiviteten biokjemisk.
- Hypoalbuminemi. Vær obs på at pasientene kan ha ødemer og samtidig redusert intravaskulært volum. I slike situasjoner kan diuretika være risikabelt.
- Hypertriglyseridemi. Det er ikke anbefalt å måle triglyserider rutinemessig. Det er ikke dokumentert at spesifikk behandling bør gis, eller at man trenger å dosejustere asparaginase (Burke et al., 2020).
Høydose metotrexat
Sist faglig oppdatert: 21.12.2023
Pasientene bør være godt hydrert fra før oppstart og fram til metotrexat er skilt ut (konsentrasjon <0.2 umol/l). Husk at metotrexat ikke kan startes før urinen er alkalisk, og at urinen bør holdes alkalisk helt til metotrexat er skilt ut. Calciumfolinat motgift er helt essensielt, og det er svært viktig at dosene justeres etter metotrexat-nivåene i serum, se tabell nedenfor. Husk å seponere alle medikamenter som kan interagere.
Interaksjoner
Tyrosin kinase hemmere inkludert imatinib, protonpumpehemmere, trimetoprim, sulfonamider (i A2G er det ikke lenger anbefalt å nulle profylakse-dose bactrim før/under HD MTX)
NSAID, salicyl, tetracykliner, phenytoin, penicillin, teofylin, samt alle medikamenter som påvirker GFR.
Dosereduksjon
Hvis man ved forrige HD MTX opplevde uttalt benmargssvikt kan man redusere mercaptopurin-dosen som gis samtidig med HD MTX fra en uke før og til en uke etter.
Hvis man ved forrige HD MTX opplevde problematisk forsinket utskillelse (f. eks nyresvikt eller uttalt mukositt) kan neste dose reduseres.
A2G har følgende retnigslinjer: “No MTX dose reduction is routinely indicated in case of prior myelotoxicity or mucositis. In case of acute kidney injury (AKI) (>50% rise in s-creatinine) within the first 1-2 days of the previous HD- MTX, the next HD-MTX can be given at full dosage, if the se-creatinine at the start of the next HD-MX has normalized.”
Ved redusert nyrefunksjon ved oppstart bør dosen reduseres.
Fedme: Metotrexat distribueres ikke til fettvev, og maks overflatebegrensning 2.2 m2 bør følges.
Forsiktighet ved pleura eller ascitesvæske (akkumulering av metotrexat).
Bivirkninger
Slimhinnetoksistet, nyresvikt, leverpåvirkning, pneumonitt. Akutt metotrexat encephalopati er et syndrom med nevrologiske utfall som oppstår innen to uker etter iv eller it behandling. Tilstanden er vanligvis fullt reversibelt, og metotrexat kan forsøkes gjentatt senere hvis reaksjonen ikke var livstruende.
Vanligvis lite kvalme og lite benmargshemming.
Antidot
MOTGIFT MED KALSIUMFOLINAT MÅ ALDRI GLEMMES!
Kalsiumfolinat (rescovolin/leukovorin) startes 42 timer etter MTX start. Første dose gis i dosering 15 mg/m2. Det skal gis en ekstradose så snart man har svaret på 42 timers verdien hvis s-MTX da er >1.0 umol/l. Dosering på denne ekstradosen er som følger:
1.0-1.9 umol/l: Calsiumfolinat-dose 15 mg/m2
2.0-2.9 umol/l: Calsiumfolinat-dose 30 mg/m2
3.0-3.9 umol/l: Calsiumfolinat-dose 45 mg/m2
4.0-4.9 umol/l: Calsiumfolinat-dose 60 mg/m2
5-6 umol/l: 75 mg/m2
6-7 umol/ : 90 mg/m2
7-8 umol/l: 105 mg/m2
8-9 umol/l: 120 mg/m2
9-10 umol/l: 135 mg/m2
>10 umol/l: 150 mg/m2
Fra og med 48 t etter start av MTX skal Leukovorin gis hver 6. time og doseres etter siste MTX konsentrasjon etter følgende skjema:
<0.2 umol/l: Calsiumfolinat avsluttes
0.2-1.0 umol/l: Calciumfolinat-dose 15 mg/m2
1.0-1.9 umol/l: Calsiumfolinat-dose 30 mg/m2
2.0-2.9 umol/l: Calsiumfolinat-dose 45 mg/m2
3.0-3.9 umol/l: Calsiumfolinat-dose 60 mg/m2
4.0-4.9 umol/l: Calsiumfolinat-dose 75 mg/m2
5-6 umol/l: 90 mg/m2
6-7 umol/ : 105 mg/m2
7-8 umol/l: 120 mg/m2
8-9 umol/l: 135 mg/m2
9-10 umol/l: 150 mg/m2
10-20 umol/l: 225 mg/m2
20-30 umol/l: 375 mg/m2
>30 umol/l: 525 mg/m2
Calsiumfolinat skal ikke gis raskere enn maksimalt 160 mg/min pga fare for hyperkalsemi. Kalsium måles i serum/plasma hver time ved doser høyere enn 375 mg/m2. Ekstra væske og bikarbonat gis ved forsinket utskillelse (min 2,5 l/m2/d, væske/bikarbonat-opplegg på dag 1 og 2 kan kopieres til dag 3 og utover).
Ved dobling av kreatinin etter 24 timer: Vurder å starte Leukovorin med en gang. Man kan da gi 525 mg/m2 hver 6. time ved nivåer av MTX over 30 umol/l, og ellers følge dosering som angitt over.
Kontroll
Sist faglig oppdatert: 21.12.2023
Det henvises til NOPHO 2008 og A2G protokollen for tidspunkt for benmarg/MRD målinger. Under vedlikeholdsbehandlingen bør pasientene kontrolleres hver 2.–3. uke med måling av hematologi og lever/galle/nyre/pankreas (hvis asparaginase) prøver.
Hos MRD-negative pasienter med donor hvor man velger å ikke transplantere i KR1, og hvor det var høyrisikofaktorer til stede ved diagnosetidspunkt, bør man måle MRD hver 3. måned det første året etter avsluttet konsolidering.
Etter avsluttet behandling bør man ved B-ALL gjøre månedlige kontroller det første året med rutinehematologiske blodprøver, deretter kontroll hver 3. måned i to påfølgende år. Ved T-ALL kan man avslutte kontrollene 3 måneder etter avsluttet behandling. Det anbefales ikke rutineprøve av beinmarg eller spinalvæske, og heller ikke rutine radiologiske undersøkelser etter avsluttet behandling. Egne regler gjelder for A2G/NOPHO 2008.
Registrering
Sist faglig oppdatert: 21.12.2023
Pasientene skal registreres i Norsk Kreftregister.
Pasienter i A2G (16-45 år) registreres i tillegg både i Castor og NOPHO databasen.
Pasienter med Ph neg ALL og alder 46–65 registreres i tillegg i NOPHO 2008 databasen, mens pasienter >65 år registreres i Web CRF3 til Eldreprotokollen.
Alle andre pasienter bør også registreres prospektivt i Web CRF3 (https://webcrf3.medisin.ntnu.no).
Det må innhentes samtykke for registrering i Web CRF3 (eget samtykket for Eldreprotokollen, og ett for øvrige pasienter (se vedlegg)). Kontakt petter.quist-paulsen@stolav.no for brukernavn/passord.
Vedlegg
Sist faglig oppdatert: 21.12.2023
1) Behandlingsoversikt for bruk av blinatumomab.
2) HyperCVAD del 1 og 2
3) HyperCVAD vedlikehold (POMP)
4) Palliativ ALL-behandling: OPAL kur
5) GM-ALL B-ALL/NHL2002 for Burkitt Lymfom/leukemi
1) Behandlingsoversikt for bruk av blinatumomab.
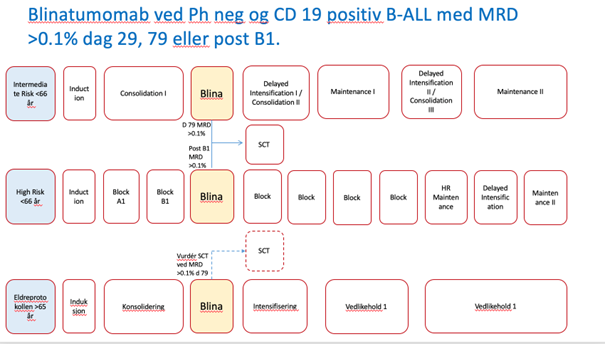
2) HYPER-CVAD del 1
1. del (HyperCVAD) og 2. del (Metotrexate-AraC) gis alternerende i alt 8 ganger (4 av hver type kur), etterfulgt av vedlikeholdsbehandling. Neste kur startes ved hvite >3 og trc>60 x 10 9/l. Dosereduksjoner ved hyperbilirubinemi, kreatininstigning, alder >60 eller alvorlig toksisitet i tidligere kur. Originalprotokoll bruker G-CSF fra dag 5 til granulocytt-regenerasjon, og oral profylakse med ciprofloxacin, flukonazol og acyclovir.
Ved Philadelphia-kromosom-positiv ALL gis Imatinib 400–600 mg /dag kontinuerlig fra diagnosetidspunkt.
Ved CD20 positiv sykdom (>20 %) gis Rituximab 375 mg/m2 iv dag 1 og 11 av Hyper CVAD og dag 1 og 8 ved Metorexat-Ara-C ved de fire første syklusene, til sammen 8 doser.
1. blokk: Hyper CVAD
Dag 1 | Dag 2 | Dag 3 | Dag 4 | Dag 8 | Dag 11 | Dag 12–14 |
|---|---|---|---|---|---|---|
Cyklofosfamid 300 mg/m2 iv over 3 timer x 2 (hver 12. time) | Cyklofosfamid 300 mg/ m2 iv over 3 timer x 2 (hver 12. time) | Cyklofosfamid 300 mg/ m2iv over 3 timer x 2 (hver 12. time) | Vinkristin 2 mg iv |
| Vinkristin 2 mg iv Evt Rituximab |
|
Mesna 300 mg iv/ m2 x 2 kontinuerlig inf Start 1t før cyklofosfamid, til 6 timer etter siste dose | Mesna 300 mg iv/ m2 x 2 kontinuerlig inf. Start 1t før cyklofosfamid, til 6 timer etter siste dose | Mesna 300 mg iv/ m2x 2 kontinuerlig inf. Start 1 t før cyklofosfamid, til 6 timer etter siste dose |
|
|
|
|
Evt Rituximab |
|
| Doxorubicin 50 mg/ m2 iv/2t |
|
|
|
Dexamethason | Dexamethason | Dexamethason | Dexamethason |
| Dexamethason | Dexamethason 40 mg x1 po |
| Metotrexat 12 mg it |
|
| Ara-C 100 mg it |
|
|
Hyper-CVAD del 2
Dag 1 | Dag 2 | Dag 3 | Dag 8 |
|---|---|---|---|
Metotrexat 200 mg/ m2iv over 2 timer fulgt av 800 mg/m2 iv over 22 timer1 |
| Folininsyre 15 mg iv hver 6. time, 8 doser. Start 36 timer etter start av MTX-infusjon2 | Evt Rituximab |
Evt Rituximab | Ara-C 3 g/m2over 2 timer x2 (hver 12. time)3 | Ara-C 3 g/m2 over 2 timer x2 (hver 12. time)3 |
|
Metylprednisolon 50 mg iv x2 | Metylprednisolon 50 mg iv x2 | Metylprednisolon 50 mg iv x2 |
|
| Metotrexat 12 mg it |
| Ara-C 100 mg it |
Dosereduksjoner
1: Metotrexat reduseres med 25 % hvis kreatinin er mellom 130 og 170 umol/l, med 50 % for høyere nivåer.
2: Doseøkning av folinsyre ved forsinket eliminering: se egen Mtx instruks.
3: Ved alder over 60 år: Metotrexat reduseres til 500 mg/m2, Ara-C til 1 g/m2 , Cyklofosfamid til 250 mg/m2, doxorubicin til 37.5 mg/m2 over 24 t og dexametason til 20 mg dag 1-4 og 11-14.
4: Hyperbilirubinemi: Vinkristin 1 mg ved bilirubin>34 umol/l, doxorubicin reduseres med 25 % ved bilirubin 34–51 mmol/l, 50 % ved 51–68 mmol/l, og med 75 % ved høyere verdier.
5: Ara-C dosereduseres til 1 g/m2 Ved kreatinin >170 umol/l, eller forsinket Mtx-utskillelse i tidligere kur. Alvorlig toksisitet i tidligere kur: Vurder 25–50 % reduksjon særlig av Mtx og Ara-C.
3) Hyper CVAD Vedlikehold (POMP)
1. Kontinuerlig peroral behandling med
- 6-Mercaptopurin (Puri-Nethol) tbl 50–150 mg x 1 po/ dag. Tas om kvelden utenom melkeprodukter.
- Metotrexat tbl 20 mg/m2 po én dag hver uke
Det anbefales å starte med 6-MP og finne tolerabel dose over 2–3 uker før metotrexat legges til en dag/uke.
Det tilstrebes moderat beinmargshemning (neutrofile 0,5–1,5x 109/l). Ved problemer anbefales å redusere metotrexat først.
Ved større neutropeniproblemer bør pasientens TPMT aktivitet bestemmes (hereditær polymorfisme).
Ved behandlingstrengende infeksjoner bør behandlingsintensiteten dempes noe. Metotrexatdosen reduseres ved plagsom munnsårhet. Cave allopurinol (interaksjon med merkaptopurin).
2. En gang/mnd
- Vinkristin 2 mg iv,
- Prednisolon 200 mg po/dag i 5 dager
Vinkristin seponeres ved pareser. Steroiddosen reduseres ved høy alder/komorbiditet/intoleranse. Vedlikeholdsbehandlingen avsluttes etter to år.
4) Palliativ ALL-behandling: OPAL kur
- Vinkristin 2 mg iv dag 1
- Prednisolon tbl 50 mg x 4 dag 1–6
- Doxorubicin 40 mg iv dag 1
- Pegylert Asparaginase 1000 IE/m2 i.m. dag 1
Kurene gis med ca 3 ukers mellomrom. Ved oppnådd maksimaldose antracyklin eller ved redusert ejeksjonsfraksjon kan doxorubicin erstattes med cytarabin 100 mg sc dag 1–6. Etter oppnådd remisjon kan man gå over til POMP vedlikehold.
5) GM-B-ALL 2002. Behandling av Burkitt lymfom/leukemi.
Se undertekst for dosejustering ved alder >55 år.
.png)
Myeloproliferative neoplasier (MPN)
Sist faglig oppdatert: 21.12.2023
Dette kapittelet omtaler diagnostikk og behandling av polycytemia vera, essensiell trombocytose, prefibrotisk myelofibrose, primær myelofibrose, sekundær myelofibrose og uklassifiserbare myeloproliferative tilstander.
Mens kronisk myelogen leukemi er omtalt i eget kapittel, omtales ikke kronisk nøytrofil leukemi og kronisk eosinofil leukemi i handlingsprogrammet for maligne blodsykdommer
Kapittelet er oppdatert i henhold til siste 5. utgave av WHO klassifikasjon (2022) for diagnostikk. I tillegg til 5. utgave av WHO klassifikasjonen foreligger det fra 2022 også et alternativt klassifikasjonssystem, International Consensus Classification (ICC). For de myeloproliferative sykdommene er det ingen forskjeller mellom WHO og ICC er klassifikasjonene, og nytt klassifiseringssystem har derfor ingen betydning for dette handlingsprogrammet.
Fortsatt er det viktig å bruke de diagnostisk kriteriene til diagnostikken av de ulike diagnosene, både hoved- og bikriterier.
Kapittelet er også oppdatert ihht. ny retningslinjer for risikostratifisering og behandling av MPN.
Introduksjon
Sist faglig oppdatert: 21.12.2023
De myeloproliferative neoplasiene (MPN) polycytemia vera (PV), essensiell trombocytose (ET), prefibrotisk myelofibrose (prePMF) og primær myelofibrose (PMF) er karakterisert ved klonal ekspansjon av erytropoiese, granulopoiese eller trombopoiese, alene eller i kombinasjon. Sykdommene medfører i økt risiko for arterielle og venøse tromboser hos de fleste men også utvikling av beinmargsfibrose, splenomegali, ekstramedullær hematopoiese og transformasjon til akutt myelogen leukemi hos en mindre andel av pasientene. Det er stor variasjon mellom de tre sykdommene med lavest risiko for transformasjon ved essensiell trombocytose og høyest ved myelofibrose. Risiko for trombose er størst ved polycytemia vera. Sykdommene er også ledsaget av kronisk inflammasjon med varierende symptomatologi som hos noen pasienter gir betydelig nedsatt livskvalitet.
Ved PV, ET og MF påvises det hos nesten alle pasientene en drivermutasjon i en av følgende tre gener: Janus kinase 2 (JAK2), Calreticulin (CALR) eller trombopoietin reseptor (MPL), alle genene aktiverer JAK-STAT signalering. Tilstedeværelse av en av drivermutasjonene utelukker nesten alltid mutasjon i en av de andre drivermutasjoner. Mutasjonene har gitt bedre forståelse for de overlappende kliniske trekk ved kronisk MPN og er sentrale i diagnostikk.
Hos noen pasienter med klinisk bilde og funn i blod og benmarg som ved myeloproliferativ sykdom men påvises ingen drivermutasjon. Disse pasientene kalles trippelnegative. Trippelnegativ myelofibrose har høyere risiko for progresjon til akutt leukemi sammenlignet med MF med drivermutasjonene.
Tabellen nr 1 viser omtrentlig forekomst av drivermutasjone ved de forskjellige myeloprofilirative tilstandene:
| Neoplasi | |||
|---|---|---|---|---|
Mutasjon | PV | ET | Pre-MF | MF |
JAK2V617F | 98% | 55% | 55% | 60% |
JAK2 exon 12 | 2 % | 0% | 0% |
|
CALR | 0% | 30% | 30% | 20% |
MPL W515 | 0% | 5% | 5% | 10% |
Trippel negativ | 2% | 10% | 10% | 10% |
Ved PV påvises JAK2V617F hos 98% av pasientene, mens mutasjon i JAK2 exon12 påvises hos noen få pasienter. JAK2 exon 12 mutasjon er assosiert med en litt annen klinisk fenotype med isolert erytrocytose i motsetning til JAK2V617F mutert PV som hovedregel har trilinjær hyperplasi/cytose.
Ved ET vil JAK2 mutasjon ofte være assosiert med høy Hb og høye hvite, høyere forekomst av trombose og økt tendens til transformasjon. CARL mutasjon ved ET ses ofte hos yngre og med høye platetall, men mindre risiko for trombose.
Vanligste mutasjoner i CALR genet er enten: type 1 mutasjon (en 52 basepar delesjon); type 2 mutasjon (5 base par insersjon). Andre mutasjonene benevnes som type 1-lignende/ type-2 lignende etter hvilke endringer det medfører i proteinet.
Mutasjon i CALR påvirker klinisk fenotype ved ET og PMF. Ved PMF er CALR type 1 mutasjon assosiert med redusert risiko for transformasjon og derav bedre overlevelse. Analyserende laboratorium bør derfor angi om det foreligger en type 1 eller type 2 CALR mutasjon.
Myeloid mutasjonspanel og cytogenetikk anbefales ikke rutinemessig, men gjøres i følgende situasjoner:
Hos pasienter med MF der allogen stamcelletransplantasjon kan være aktuelt. Hos allo-SCT kandidater vil myeloid panel og cytogenetikk ha viktig prognostisk betydning.
Ved fravær av ovennevnte drivermutasjoner og usikkerhet om det foreligger klonal (MPN) sykdom kan myeloidpanel/cytogenetikk være avklarende.
Diagnostikk ved MPN
Sist faglig oppdatert: 21.12.2023
- MPN diagnostiseres i henhold til 5. utgave av WHO klassifikasjonen ved hjelp av diagnostisk kriterier som er beskrevet nedenfor.
- Den diagnostiske utredningen ved MPN krever
- Blodprøver.
- Mutasjonsanalyser: JAK2 (V617F eventuelt exon12 mutasjon), MPL og CALR.
- Benmargsbiopsi. Hos pasienter med helt sikre tegn på PV kan benmargsbiopsi utelates.
Essensiell trombocytose
Diagnosen ET krever:
- Alle fire hovedkriteriene.
- Eller de første tre hovedkriteriene og minst ett bikriterium.
Hovedkriterier
- Vedvarende blodplatetall ≥ 450 x 109/L.
- Beinmargsbiopsi funn (obligatorisk, for å kunne skille mellom ET og prefibrotisk myelofibrose) med overveiende proliferasjon av megakaryocytter med økt antall forstørrede, modne megakaryocytter med hypersegmenterte kjerner. Ingen vesentlig økning eller venstreforskyvning av granulopoiese eller erytropoiese, og veldig sjelden en liten økning i retikulære fibre, ikke høyere enn europeisk MF grad 1 (fibrosen graderes 0-3).
- Tilfredsstiller ikke WHO kriteriene for KML, PV, PMF, MDS eller andre myeloide neoplasier.
- Tilstedeværelse av JAK2, CALR eller MPL mutasjon.
Bikriterium
- Ingen holdepunkt for reaktiv trombocytose
- Tilstedeværelse av en klonal markør (annet enn en drivermutasjon).
Polycytemia vera
Diagnosen PV krever:
- Alle tre hovedkriteriene er oppfylt.
- eller at de to første hovedkriteriene samt at bikriteriet er oppfylt.
Hovedkriterier:
- Hemoglobin > 16,5 g/dL hos menn, > 16,0 g/dL hos kvinner,
eller Hct > 49 % hos menn, Hct > 48 % hos kvinner - Beinmargsbiopsi* som viser hypercellularitet i forhold til alder og med trilineær proliferasjon (panmyelose) inkludert framtredende erytroid-, granulocytt- og megakaryocyttproliferasjon med pleomorfe, modne megakaryocytter.
- Påvisning av JAK2 V617F eller JAK2 exon 12 mutasjon.
Bikriterium:
1. Subnormalt serum EPO nivå.
Beinmargsbiopsikriteriet er ikke nødvendig i tilfeller med vedvarende absolutt erytrocytose: hemoglobin >18,5 g/dL hos menn (hematokrit 55,5 %) eller >16,5 g/dL hos kvinner (hematokrit 49,5 %) hvis hovedkriterium 3 og bikriteriet er oppfylt. Benmargsbiopsi er viktig å gjennomføre dersom hovedkriterium 3 er fraværende, men bikriteriet er tilstede.
Polycytemia vera har sannsynligvis vært underdiagnostisert tidligere pga høye Hb/Hct krav i WHO 2008. Såkalt maskert PV (for eksempel pga jernmangel) har ofte ikke blitt diagnostisert eller klinisk blitt tolket som ET. Siden 2016 ble grensen for Hb/Hct grensene senket, og benmargsbiopsikriteriet ble oppgradert fra bikriterium til hovedkriterium. Supprimert EPO er et tegn på PV og kan bidra til å støtte diagnosen (maskert) PV.
Prefibrotisk myelofibrose
Diagnosen pre-MF krever:
- Alle tre hovedkriteriene samt tilstedeværelse av minst ett bikriterium.
Hovedkriterier:
- Beinmargsbiopsi viser megakaryocytt proliferasjon og atypi, uten retikulin fibrose > MF grad 1 (fibrosen graderes 0-3), ledsaget av økt alders-justert benmargs cellularitet, granulocytt proliferasjon og ofte redusert erytropoiese.
- Tilfredsstiller ikke WHO kriteriene for KML, PV, ET, MDS, eller andre myeloide neoplasier.
- Tilstedeværelse av enten
- JAK2, CALR or MPL mutasjon,
eller
- JAK2, CALR or MPL mutasjon,
-
- Tilstedeværelse av annen typisk myelood klonal markør
eller
- Tilstedeværelse av annen typisk myelood klonal markør
-
- Fravær reaktiv retikulin benmargsfibrose
Bikriterier:
Tilstedeværelse av minst et av de følgende kriterier ved minst to anledninger:
- Anemi, ikke betinget i annen sykdom
- Leukocytose ≥ 11 x 109/L
- Splenomegali påvist ved palpasjon eller billeddiagnostikk
- LDH over øvre normalgrense
- Leukoerytroblastisk blodbilde
*ved fravær av en av drivermutasjonene, kan NGS/myleoid panel med funn av ledsagende mutasjon evt andre mutasjoner i JAK2, MPL, CALR (sjelden) være til hjelp i å avklare om det foreligger en klonal sykdom.
** mindre (grad 1) retikulin fibrose sekundær til infeksjon, autoimmun sykdom eller annen kronisk inflammatorisk tilstand, hårcelle leukemi, eller annen lymfoid neoplasi, metastatisk malignitet eller toksisk (kronisk) myelopati.
Primær myelofibrose
Diagnosen PMF krever:
- Alle tre hovedkriteriene samt tilstedeværelse av minst ett bikriterium.
Hovedkriterier
- Beinmargsbiopsi som viser megakaryocyttproliferasjon/atypi og granulocyttproliferasjon ledsaget av enten retikulin og/eller kollagen fibrose tilsvarende grad 2 eller 3 (fibrosen graderes 0-3).
- Tilfredsstiller ikke WHO kriteriene for ET, PV, KML, MDS, eller andre myeloide neoplasier.
- Påvisning av enten
- JAK2, CALR or MPL mutasjon,
eller
- JAK2, CALR or MPL mutasjon,
-
- Tilstedeværelse av annen typisk myelood klonal markør
eller
- Tilstedeværelse av annen typisk myelood klonal markør
-
- fravær av reaktiv retikulin benmargsfibrose **.
Bikriterier:
- Anemi, ikke er betinget i annen sykdom
- Leukocytose ≥ 11 x 109/L
- Palpabel splenomegali
- Serum LD økning over øvre normalgrense for angjeldende laboratorium
- Leukoerytoblastose i perifert blodutstryk
*ved fravær av en av driver-mutasjonene, kan NGS/myleoid panel med funn av ledsagende mutasjon (f. eks. ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1) evt andre mutasjoner i JAK2, MPL, CALR (sjelden) være til hjelp i å avklare om det foreligger en klonal sykdom.
**Benmargsfibrose sekundær til infeksjon, autoimmun sykdom eller annen kronisk inflammatorisk tilstand, hårcelle leukemi eller annen lymfoid malignitet, metastatisk malignitet eller toksisk (kronisk) myelopati.
Uklassifiserbar MPN (U-MPN)
- Diagnosen brukes når biopsi, laboratorieprøver, molekylærpatologi og kliniske opplysninger taler for MPN sykdom, men subklassifisering ikke er mulig i hht WHO diagnostiske kriterier. Gjentagelse av biopsi bør vurderes ved oppfølging.
Risikoklassifisering og behandlingsanbefalinger
Sist faglig oppdatert: 21.12.2023
Alle pasienten med MPN skal risiko stratifiseres før oppstart av behandling.
- For ET og PV risikostratifiseres pasientene mtp. trombose/blødnings risiko.
- For MF risikoklassifiseres pasientene etter risiko for overgang til sekundær AML og utvikling av beinmargssvikt.
Risikostratifisering og behandling av essensiell trombocytose
Risikoklassifiseringen ved ET predikerer først og fremst risikoen for tromboemboliske komplikasjoner. I Norge anbefales å bruke International Prognostic Score (IPSET)
der pasientene kategoriseres i en av følgende 4 risikogrupper: veldig lav, lav, intermediær og høyrisiko.
Veldig lav risiko: fravær av tidligere trombose, alder < 60 år, JAK2 negativ.
Lav risiko: fravær av tidligere trombose, alder < 60 år, JAK2 positiv.
Intermediær: fravær av tidligere trombose, alder > 60 år, JAK-2 negativ.
Høy risiko: tidligere trombose eller alder > 60 år og JAK2 positiv.
Behandlingsmålet er å redusere både arterielle og venøse trombotiske komplikasjoner, unngå blødning, samt å redusere symptomene. Pasienter med ET har nærmest lik overlevelse som normalbefolkningen. Hos noen få pasienter ser man, typisk mange år etter diagnose, overgang til sekundær MF (post-ET MF) og evt sekundær AML, men risiko er betraktelig lavere enn ved PV.
Følgende behandling anbefales
- Albyl E 75 mg (ASA) x 1 daglig anbefales til alle med unntak av pasienter med veldig lavrisiko pasienter uten hjerte-kar risiko. Man bør også være tilbakeholdende med ASA hos personer med CALR mutasjon uten hjerte-kar risiko og alder < 60 år grunnet risiko for blødning (Alvarez-Larrán et al., 2021).
- Albyl-E 75mg x 2 kan vurderes til høy risikopasienter og til pasienter med vedvarende mikrovaskulære symptomer som allerede bruker Albyl-E 75 mg x 1 og cytoreduktiv behandling.
- Albyl-E bør ikke gis ved blodplatetall > 1500 x109/L pga økt blødningstendens, og med forsiktighet ved >1000 x109/L (evidensgrad B ) mulig som følge av økt forbruk av von Wllibrand faktor.
- Albyl E anbefales til pasienter som har hatt blodplatetall >1000 x109/L etter blodplatetallet er redusert med cytoreduktiv behandling.
- Albyl E kan vurderes til høyrisiko pasienter med hjerte-kar risiko som har gjennomgått venøs trombose og som bruker annen antikoagulasjonsbehandling.
- Blødningsfare må vurderes og det anbefales ved samtidig indikasjon for DOAK at lavest mulig dosering av DOAK benyttes.
- Cytoreduktiv behandling anbefales til intermediær og høyrisiko pasienter og til lav risiko pasienter med mikrovaskulære symptomer, JAK2 mutasjon, kardiovaskulære risikofaktorer eller ved leukocytose (>15 x 109/L).
- Blodplater bør reduseres til < 400 x 109/L.
- Cytoreduktiv behandling bør gis ved blodplatetall > 1500 x 109/L, også hos lavrisiko, pga. økt risiko for blødning.
- Pasienter < 60-65 år: interferon-a som første valg, hydroxyurea eller anagrelid som andre valg (evidensgrad A) (Birgegård et al., 2018; Gisslinger et al., 2013).
- Pasienter > 60-65 år: hydroxyurea som første valg, interferon-a eller anagrelide som andrevalg (evidensgrad A) (Landtblom et al., 2018).
- Intermitterende busulfan tabletter er tredjelinjevalg hos eldre pasienter som er resistent eller intolerant for annen behandling.
- Hydroxyurea er det best dokumenterte behandlingsvalg ved ET, og er i en studie like god som anagrelide i fase 3 studie (Gisslinger et al., 2013). I en studie ble det ved bruk av anagrelide observert økt risiko for blødning, arteriell trombose og utvikling av myelofibrose.
- Det er ikke funnet noen sikker økt risiko for sekundær AML ved bruk av hydroxyurea alene, men derimot mulig økt risiko for ulike typer hudkreft. Grunnet lang eksponeringstid ved bruk fra ung alder anbefales tilbakeholdenhet med bruk av hydroxyurea hos pasienter < 60-65 år).
- Hydroxyurea må ikke brukes ved graviditet eller om graviditet planlegges.
Risikostratifisering og behandling av Polycytemia vera
Risikoklassifiseringen ved PV vurderer risiko mtp. trombotiske komplikasjoner.
Høyrisikogruppe: alder >60 år og/eller tidligere trombose.
Lavrisikogruppe: alder <60 år uten tidligere trombose.
Behandlingsmålet er primært å redusere risikoen for trombotiske komplikasjoner og redusere symptomer. Pasienter med PV har økt risiko for transformasjon til sekundær MF (post-PV MF) og og høyere riskio enn ved ET. Behandlingsrådene har også som mål å minimalisere risikoen for senere overgang til post-PV myelofibrose og/eller akutt leukemi. Ved transformasjon er behandling og oppfølging som beskrevet i kapittel om PMF.
- Venesectio anbefales for å holde hematokrit < 45%.
- Oppstart raskt ved mistanke om PV, altså før svar på diagnostikk foreligger, spesielt ved uttalt erytrocytose.
- Albyl-E 75 mg x 1 daglig med mindre det foreligger kontraindikasjoner. Alternativt klopidogrel dersom pasienten har intoleranse for Albyl-E.
- Albyl-E 75mg x 2 kan gis til pasienter med vedvarende plagsomme mikrovaskulære symptomer som ikke responderer på Albyl-E 75 mg x 1 og cytoreduktiv behandling
- Cytoreduksjon anbefales til høyrisiko pasienter. Behandlingsmål er hct < 0,45 og bedring/normalisering av trombocytter og leukocytter.
- Ved lav risiko sykdom bør cytoreduksjon også vurderes ved dårlig toleranse/ikke optimal effekt av venesectio, leukocytose >15 x 109/L, symptomatisk eller progressiv splenomegali, systemiske symptomer.
- Anbefalt cytoreduktiv behandling:
- < 60-65 år: førstevalg: interferon-a; andrevalg: hydroxyurea eller ruxolitinib.
- > 60-65 år: førstevalg hydroxyurea; andrevalg: interferon-a eller ruxolitinib.
- Hos unge passienter med behov for raskt innsettende effekt velges hydroxyurea og så overgang til interferon-alfa.
- Kombinasjonsbehandling (hydroxyurea-anagrelide, hydroxyurea- interferon-alfa, interferon-alfa + anagrelide) kan være aktuelt hos pasienter med utilstrekkelig effekt av et preparat eller intoleranse.
- Intermitterende busulfan tbl kan anvendes hos eldre som har intoleranse for annen behandling
Risikoklassifisering og behandling ved myelofibrose: prefibrotisk, primær og sekundær
Prefibrotisk myelofibrose:
Pasientene bør primært risikostratifiseres som ET og bruk av Albyl-E og cytoreduktiv behandling følger de samme retningslinjer som ved ET (Guglielmelli et al., 2020).
Ved overgang til myelofibrose, risikostratifieres og behandles som for PMF.
Primær/sekundær myelofibrose:
Forventet overlevelse ved myelofibrose er kortere enn ved essensiell trombocytose og poycytemia vera. Risikoklassifiseringen ved myelofibrose er relatert til risiko for transformasjon og overlevelse.
Den eneste mulige kurative behandlingen er allogen stamcelletransplantasjon. Allogen stamcelletransplantasjon er assosiert med en ikke ubetydelig risiko for behandlingsrelaterte komplikasjoner og tilbys kun pasienter med lav grad av komorbiditet med stor risiko for transformasjon.
Primær myelofibrose risikostratifiseres tradisjonelt med IPSS ved diagnose og DIPSS+ ved senere oppfølging. MYSEC-PM benytte til å risikostratifisere post-ET og post-PV.
Høyrisiko mutasjoner/cytogenetikk er inkludert i nye prognostiske systemer som MIPSS70+ og GIPSS og anvendes nå rutinemessig, særlig ved oppfølging/vurdering av pasienter som er transplantasjonskandidater.
Hos de fleste pasientene vil behandlingen være symptomrettet og palliativ, og må ta utgangspunkt i de aktuelle kliniske problemer. Eneste unntak kan være interferon-alfa som kan ha sykdomsmodifiserende effekt i tidlig stadium av MF, bla med reduksjon av JAK2/CALR/MPL allelbyrde.
Over 25% av MF pasientene vil før eller senere transformere til blastfase, dvs. sekundær AML, med ≥20% blaster i blod/benmarg. Disse pasientene vil typisk gå via en aksellert fase (10-19% blaster i blod/benmarg). Ofte vil blastandel være lettere å vurdere, og mer representativt, i perifert blodutstryk enn i benmarg, dels pga. uttalt ekstramedullær sykdom.
I de prognostiske systemene gir blastandel >1-2% i perifert blod økt risiko for sekundær AML/død. Blodutstryk for å fange opp økende blastandel i blod er derfor et viktig verktøy i oppfølging/klassifisering av MF pasienter.
Henvisning allogen stamcelletransplantasjon eller symptomatisk behandling:
Ved PMF/sekundær MF er det konsensus å henvise til vurdering for allotransplantasjon hos pasienter med akseptabel komorbiditet hvis leveutsiktene i risikoscore er mindre enn 5 år vurdert ut fra ovennevnte prognostiske verktøy.
Behandling av hyperproliferasjon, splenomegali og konstitusjonelle symptomer:
Interferon-a anbefales som førstelinjebehandling hos yngre pasienter (<60 år) som ikke er umiddelbare kandidater for transplantasjon. Pasienten bør være i den tidlige proliferative fase av sykdommen uten avansert fibrose eller splenomegali
Hydroxyurea er første valg hos eldre pasienter over 60-65 år ved symptomatisk splenomegali, konstitusjonelle symptomer og for å kontrollere trombocytose og leukocytose.
Hydroxyurea og interferon-alfa har ofte begrenset effekt ved avansert sykdom med betydelig splenomegali og fibrose og bør helst brukes i tidlig stadium av MF.
JAK hemmere (fedratinib, ruxolitinib) brukes ved symptomatisk splenomegali eller konstitusjonelle symptomer hos pasienter som ikke har tilstrekkelig nytte/toleranse for hydroxyurea eller interferon (evidensgrad A). Mange bruker også JAK hemmer som første linje behandling ved høyrisiko/uttalt splenomegali/symptombyrde. Man bør derimot være oppmerksom på at god dokumentasjon på at JAK-hemming bedrer pognose/forsinker sykdomsutvikling er mangelfull. I tilegg vil JAK hemming hos de fleste pasienter kunne foverre anemi og på lang sikt øke infeksjonsfare. Den kliniske nytte av JAK hemmer bør vurderes fortløpende. Hos de med suboptimale response på splenomegali med ruxolitinib har studien vist at 30% responderer på fedratinib som 2.linjesbehandling.
Alternativ cytostatika/cytoreduktiv behandling kan ha effekt ved hydroxyurea resistens, f. eks busulfan og anagrelide (ekstrem trombocytose).
Strålebehandling av milt kan gi symptomatisk bedring, men effekten varer kun 3-6 måneder. Strålebehandling kan også anvendes mot annen symptomgivende ekstramedullær hematopoiese og har nok en større rolle enn stråling av milt.
Splenektomi kan være indisert ved symptomatisk portal hypertensjon og medikament-refraktær betydelig splenomegali, men har en perioperativ mortalitet på over 10 %. Komplikasjoner oppstår hos ca 50 %. Disse pasientene bør vurderes ved universitetssykehus i forkant av kirurgi.
Avansert/akselerert fase MF definert som økende blastandel i blod/benmarg (10-19%) har dårlig prognose og fare for rask overgang til AML. Kombinasjon av hypometylerende medikament og ruxolitinib er aktuell behandling.
Anagrelide kan brukes ved uttalt trombocytose og intoleranse for andre konvensjonelle cytoreduktive medikamenter.
Busulfan tabletter kan anvendes hos eldre med hyperproliferasjon ved resistens/intoleranse for annen behandling.
Behandling av anemi ved myelofibrose:
Prednisolon anbefales ved Coombs/DAT positiv hemolytisk anemi.
Erytropoietin/ESA anbefales som førstelinjebehandling (ca 25-50% oppnår effekt), lavest hos transfusjonstrengende pasienter og høyest hos eldre pasienter og hos de med s-erythropoietin < 250. Det kan også ha en rolle ved JAK hemmer mediert anemi.
Danazol (± prednisolon), anbefales som alternativ førstelinjebehandling, ca 2/3 oppnår respons.
Lavdose thalidomide eller lenalidomid +/- prednisolon alternativ ved manglende effekt av erytropoietin eller danazol. Særlig en opsjon ved 5q-.
Effekten av behandling er forbigående og varierer mellom 1-2 år uansett valg av medikament. Vedvarende transfusjonsterapi er ofte eneste alternativ hos noen av disse pasientene. Rettningslinjer for jernkjelering er omtalt i kapittel om MDS.
Forebygging av trombose:
- Albyl-E 75 mg x 1 anbefales til MF pasienter med JAK2 mutasjon uten øsofagus varicer, alvorlig trombocytopeni eller gjentatte blødninger i muskler og ledd.
Cytoreduktiv behandling med hydroxyurea/interferon hos MF pasienter med høyrisiko for trombose som ved leukocytose over 15, tidligere trombose, trombocytose har flere kardiovaskulære risikofaktorer.
Prognostiske scoringssystemer ved primær og sekundær myelofibrose
Det er flere prognostiske scoringssystemer. Disse scoringsystemene er viktige for planlegging av behandlingen, spesielt der hvor allogen stamcelle transplantasjon kan være aktuell behandling. De ulike prognostiske verktøy kan hos en og samme pasient av og til gi noe ulike resultat og må brukes med skjønn.
De mest vanlige har vært:
- IPSS, angir prognose ved diagnose. Kan ikke benyttes gjennom behandlingsforløpet.
Dette ble så utvidet med DIPSS/DIPSS+ som vi kan bruke i videre oppfølging av sykdommen:
- https://www.mpnconnect.com/estimate-myelofibrosis-prognosis-tool.aspx
- https://qxmd.com/calculate/calculator_315/dipss-plus-score-for-prognosis-in-myelofibrosis
Nye og prognostiske system som også tar hensyn til høy risiko genmutasjoner og cytogenetiske avvik:
- MIPPS70+ http://www.mipss70score.it/
- GIPPS https://www.uptodate.com/contents/calculator-genetically-inspired-international-prognostic-scoring-system-gipss-for-primary-myelofibrosis-in-adults
Høyrisiko mutasjoner innbefatter bla følgende (vil trolig bli utvidet/revidert etter hvert): ASXL1, EZH2, SRSF2, U2AF1, IDH1/2.
Ugunstig karyotype er typisk:
kompleks karyotype, 5q-, -7/7q-, +8, inv(3), i(17q), 17p-, 12p-, 11q-, +19, +21.
Prognostisk verdi av IPSS, DIPSS, DIPSS+, MIPPS70 og GIPPS er mindre kartlagt ved sekundær myelofibrose enn MYSEC-PM http://www.mysec-pm.eu/
Den tar stilling til blant annet driver mutasjon status, men ikke cytogenetikk/myeloid panel.
Skåringssystem | Anvendbarhet | Prognostiske faktorer | Risikoskår | Risikoskår og median overlevelse (MO) (måneder)
|
|---|---|---|---|---|
IPSS (Cervantes et al., 2009)
| Ved diagnose | Alder > 65 år | 1 |
Lav risiko (skår 0), MO 135 Intermediær- 1 risiko (skår 1), MO 95 Intermediær- 2 risiko (skår 2), MO 48 Høyrisiko (skår ≥ 3), MO 27 |
Anemi (Hb< 10g/dL) | 1 | |||
Leukocytter > 25x109/L | 1 | |||
Blaster i blod ≥ 1 % | 1 | |||
Konstitusjonelle Symptomer (feber, nattesvette, vekttap) | 1 | |||
DIPSS (Passamonti et al., 2010)
| Under hele sykdomsforløpet | Alder > 65 år | 1 |
Lav risiko (skår 0), MS ikke nådd Intermediær-1 risiko (skår 1-2), MO 170 Intermediær-2 risiko (skår 3-4), MO 48 Høyrisiko (skår 5-6), MO 18 |
Anemi (Hb< 10g/dL) | 2 | |||
Leukocytter > 25x109/L | 1 | |||
Blaster i blod ≥ 1 % | 1 | |||
Konstitusjonelle Symptomer | 1 | |||
DIPSS pluss*(Gangat et al., 2011) | Under hele sykdomsforløpet
| DIPSS lav risiko | 0 |
Lavrisiko (skår 0), MO 185 Intermediær-1 risiko (skår 1), MO 78 Intermediær-2 risiko (skår2-3), MO 35 Høyrisiko (skår ≥4), MO 16 |
DIPSS intermediær-1 | 1 | |||
DIPSS intermediær-2 | 2 | |||
DIPSS høyrisiko | 3 | |||
Transfusjons-avhengighet | 1 | |||
Ugunstig cytogenetikk¶ | 1 | |||
Trombocytter < 100x109/L | 1 |
* Kalkuler først DIPSS skår, og legg deretter til skår for transfusjonsavhengighet, cytogenetikk og trombocytopeni for å kalkulere endelig DIPSS pluss skår.
¶ Prognostisk ugunstige karyotyper er kompleks karyotype eller ett eller to avvik som inkluderer +8, -7/7q-, i(17q), -5/5q-, 12p-, inv(3), eller 11q23 rearrangering.
Allogen stamcelle transplantasjon
Allotransplantasjon bør vurderes hvis leveutsiktene er mindre enn 5 år vurdert ut fra prognostisk score og det ellers ikke foreligger noen kontraindikasjoner.
Noen pasienter med gunstig risiko iht. prognostisk score kan likevel være aktuelle ved høy symptombyrde/anemi, trippel negative pasienter (negativ for JAK2, CALR, MPL), økende blastandel.
Det kan ut fra klinikk og scoringsverktøy være vanskelig å angi rett tidspunkt for transplantasjon. Aktuelle pasienter bør henvises tidlig i forløpet til norsk gruppe for stamcelletransplantasjon og samtidig utrede donorsituasjon.
Samtidig bør pasienten ikke være i en avansert fase i form av økende blastandel før oppstart kondisjonering. Transformasjon til akselerert fase (10-19% blaster i blod/benmarg) er assosiert med dårlig overlevelse, men allerede ved blastandel >5% perifert er det tegn til dårligere utfall (Kröger et al., 2023a; Kröger et al., 2023b).
Allogen stamcelletransplantsjon ved myelofibrose er assosiert med en betydelig risiko for tidlige komplikasjoner som infeksjoner, graftsvikt og veno-okkluiv sykdom. Det er ingen øvre aldersgrense, men pasienter må nøye vurderes ut fra totalbilde og kun et mindretall av pasienter over 70 år vil i praksis være tjent med transplantasjon.
Betydelig splenomegali (>20-22cm i lengste diameter) er assosiert med dårlig utfall, spesielt graftsvikt. Det er i Norge konsensus om at man ikke ønsker å transplantere pasienter med miltstørrelse over 20cm. Pasienter med milstørrelse over 20cm tilbys behandling med JAK2 hemmer (fedratinib eller ruxolitinib). Ved manglende effekt kan man vurdere splenektomi eller miltbestråling. Splenektomi er assosiert med betydelig risiko for postoperative komplikasjoner og bør kun utføres ved Rikshospitalet for pasienter fra Helse Sør-Øst, eller ved universitetsykehus i andre regioner. Miltbestråling gir enn rask men forbigående reduksjon i miltstørrelse. Etter miltbestråling sees ofte en forverring av cytopenier. Miltbestråling kan overveies hos pasienter med utilstrekkelig effekt av JAK2 hemmer og som ansees å ikke tåle splenektomi.
Behandling av MPN
Sist faglig oppdatert: 21.12.2023
Oppsummering av behandling av MPN
Behandling av ET:
- Albyl E 75 mg til alle med unntak av veldig lavrisiko pasienter og pasienter med blodplatetall >1500 x 109/L
- Indikasjon for cytoreduktiv behandling:
- Intermediær og høyrisiko pasienter
- Lavrisiko pasienter med mikrovaskulære symptomer, ved kardiovaskulære risikofaktorer, ved leukocytose (>15 x 109/L)
- Alle med platetall >1500 x 109/L
- Førstelinje cytoreduktiv behandling ved ET:
- Alder < 60-65 år: interferon-a
- Alder > 60-65 år: hydroxyurea
- Til alle - anagrelide
Behandling av PV:
- Albyl E 75 mg til alle
- Venesectio eller cytoreduktiv behandling for å holde Hct<45%
- Førstelinje cytoreduktiv behandling ved PV:
- Alder < 60-65 år: interferon-a
- Alder > 60-65 år: hydroxyurea
Behandling av myelofibrose:
- Henvising til vurdering mtp allogen stamcelletransplantasjon for pasienter med median forventet levetid kortere enn 5 år
- Førstelinjebehandling ved DIPPS lavrisiko/ intermediær med konstitusjonelle symptomer/ splenomegali
- Alder < 60-65 år: interferon-a
- Alder > 60-65 år: hydroxyurea
- Førstelinjebehandling ved DIPPS intermediær2/høyrisikosykdom med konstitusjonelle symptomer/ splenomegali
- JAK2-hemmer: fedratinib eller ruxolitinib
- Førstelinjebehandling ved anemi:
- Erytropoietinanalog eller danazol.
- Prednisolon ved DAT+ hemolytisk anemi
Råd og gjennomføring av behandling ved MPN
Hydroxyurea
Hydroxyurea er et ikke-alkylerende, uspesifikt myelosuppressivt medikament. Startdosen er 500mg 1 til to 2 ganger daglig. Hos eldre anbefales startdosen 500mg x 1. Det bør i starten foretas relativt hyppig måling av hematologiske parametere og eventuell doseøkning for å oppnå et stabilt blodplatetall i området 200 – 400 x 109/L og hematokrit <45%. Hydroxyurea behovet ligger ofte på 10-14 tabletter à 500mg per uke. Hos eldre pasienter kan man komme til målet med lavere ukedoser. Hvis det oppstår cytopeni, må dosen reduseres slik at antallet nøytrofile granulocytter ikke blir lavere enn 1,0-1,5 x 109/L. I noen tilfeller blir nøytropenien dosebegrensende, men om man likevel klarer å oppnå et blodplatetall på < 600 x 109/L, kan dette i noen tilfeller ansees tilfredsstillende. Hydroxyurea må gis daglig og kontinuerlig. Ved avbrudd i behandlingen stiger blodplatetallet i løpet av 1-2 uker. Det kan etter seponering komme en betydelig stigning av blodplatetallet og økt tromboserisiko.
Hudsår, aktinisk keratose og plateepitel forandringer er vanlig etter noen års behandling. GI plager kan ses. Feber er uvanlig, men kan være uttalt. Observasjonstudier viser mulig økt risiko for ulike typer hudkreft. Hos noen kan blodplatetallet flukturere dramatisk ved hydroxyurea behandling, og nødvendiggjøre seponering og alternativ behandling.
Hydroxyurea må ikke brukes under graviditet eller hvor graviditet planlegges, og bør seponeres minst 3 måneder før planlagt graviditet både hos kvinner og menn.
Interferon-alfa
Interferon supprimerer vekst av multipotente hematopoietiske progenitor celler. Flere ikke randomiserte studier har vist vedvarende effekt, men varierende toleranse, for medikamentet. I tillegg har man sett molekylær respons som kan gi håp om at sykdomsforløpet påvirkes.
Pegylert interferon-alfa kan doseres en gang per uke eller annen hver uke. Pegasys 135mikrogram injeksjonsvæske, oppløsning i ferdigfylt sprøyte. Sprøyten er merket med graderinger tilsvarende 135mikrogram (mcg), 90mcg og 45mcg. Den finnes i pakninger med 1, 4 eller en multipakning på 12 (2 pakker med 6) ferdigfylte sprøyter. Startdosen for pegylert interferon alfa-2a er 45mg subcutant en gang per uke, med ukentlig doseeskalering, dvs 90 neste uke før doseøkning til 135mg og eventuelt 180mg /uke hvis man ikke får tilstrekkelig hematologisk effekt. De fleste pasientene responderer på en dose mellom 90 og 135mg per uke. Full effekt forventes etter opptil tre måneder. Kan også forlenge intervallet til annen hver uke eller lenge avhengig av respons. I tilfeller der det haster med å få ned celletallet kan man starte først med hydroksyurea.
Ro-peginterferon alfa 2b har markedsføringstillatelse for PV fra EMA, men er ikke godkjent for bruk i Norge av Nye metoder.
Hvis cytoreduktiv behandling er indisert under graviditet eller når graviditet planlegges, er det interferon man kan bruke, men det anbefales ikke brukt under amming.
Tretthet, influensalignende symptomer og humørforandringer er de mest vanlige bivirkninger, men er ofte forbigående eller lar seg kupere med paracet ved influensalignende plager. Man bør være obs på symptomer på depresjon hos disponerte pasienter. Myalgi og aktivering av eksisterende eller latent autoimmun sykdom, inkludert hypo- og hyperthyreose, er ikke uvanlig. Leverenzymer og thyroideaverdier bør monitoreres under behandlingen. Man bør ta en pause fra behandlingen dersom transaminaser øker > 2 ganger normale verdier til det har normalisert seg. Så kan man kan introdusere interferon på nytt, men med lavere dose.
Anagrelid
Anagrelid har en selektiv effekt på megakaryocytter og reduserer trombocyttnivået like effektivt som hydroxyurea. Startdosen er 0,5mg x2. Det bør gjøres tellinger 1-2 ganger ukentlig de første uker, idet blodplatetallet kan falle meget raskt. Dosen kan økes etter 1-2 uker om blodplatetallet ikke faller. Ukentlig doseøkning må ikke være >0,5mg/dag, oftest vil mindre doseøkninger være mest gunstig for ikke å få for raske blodplatefall. Maksimaldosen bør ikke overstige 3mg i en enkelt dose. De fleste tolererer dosering to ganger daglig, men dosering 3-4 ganger daglig kan være nødvendig hos dem som har mye bivirkninger (hjertebank, hodepine). Kombinasjonen Anagrelid og Albyl E bør brukes med en viss forsiktighet pga noe økt risiko for blødning.
Palpitasjoner og tachycardi er de mest vanlige bivirkningene og kan behandles med en liten dose betablokker. Noen får diare (inneholder laktose) og loperamid kan eventuelt brukes. Bivirkningene er vanligst de første ukene av behandlingen. Anagrelid bør brukes med forsiktighet hos pasienter med tidligere hjertesvikt eller arytmier.
Anagrelid må ikke brukes under graviditet eller hvor graviditet planlegges, fordi det ikke finnes data om eventuell effekt på fosteret.
JAK hemmere
Ruxolitinib
Ruxolitinib var første godkjente JAK hemmer og hemmer både JAK 1 og JAK2. Ved myelofibrose er den godkjent for symptomatisk splenomegali og for konstitusjonelle symptomer. Medikamentet har effekt uavhengig av drivermutasjon og hemmer altså JAK/STAT signaleringen som er overaktiv ved alle MPN. Ruxolitinib kan ha frapperende effekt på allmensymptomer i løpet av kun får dager, mens effekt på splenomegali tar lengre tid. Man kan som regel konkludere etter 3-6 måneders behandling.
Økende cytopeni, særlig anemi, er vanlige bivirkninger.
Startdosen er 20mg 2 ganger daglig ved blodplatetall >200 x 109/L, 15mg 2 ganger daglig ved blodplatetall 100-200 x 109/L, og høyeste anbefalte startdose er 5mg 2 ganger daglig ved blodplatetall 50 - 100 x 109/L.
Pasienter med trombocytopeni bør titreres med forsiktighet. Ved anemi velger man også ofte forsiktig dosering i starten.
Ruxolitinib har også markedsføring av EMA til behandling av PV, er til vurdering hos Nye metoder.
Ved hydroxyurea-resistent polycytemia vera er startdosen ofte 10mg x 2, og doseendring avhengig av toleranse og effekt.
Bivirkninger kan komme når som helst under behandlingsforløpet, og det er nødvendig med regelmessige hematologiske tellinger og klinisk vurdering.
Økt infeksjonstendens, opportunistiske, er et problem og man må ha dette i bakhodet. Særlig dersom pasienten er immunsupprimert av andre grunner, må man være på vakt og vurdere for eksempel Herpes zoster profylakse.
Ved seponering pga manglende respons eller bivirkning bør det skje langsomt over et par uker pga enkelte rapporter om rebound effekt med sterk symptomatisk sykdom.
Fedratinib
Fedratinib er en ny godkjent JAK hemmer, som er en selektiv hemmer av JAK2. Den virker også uavhengig av type drivermutasjon. Det er avgjort at fedratinib skal være førstevalg hos MF pasienter med indikasjon splenomegali/allmensymptomer.
Studier tyder på likeverdig effekt som ruxolitinib i første linje (Harrison et al., 2020; Pardanani et al., 2015). Rundt 30%, av de som svikter på ruxolitinib kan respondere på fedratinib (Pardanani et al., 2021). Fedratinib kan også benyttes ved intoleranse for ruxolitinib.
Anbefalt dose er 400mg x 1 ved plater >50 x 10 x 109/L men kan også starte forsiktig og titrere opp. Fedratinib blokkerer opptak av tiamin og Wernickes encefalopati er rapportert hos ca. 1 % av pasientene behandlet med Fedratinib. Det skal utføres tester av tiaminnivåer, fullstendig blodtelling, leververdier, amylase/lipase, ura og kreatinin ved baseline før behandlingsstart, regelmessig under behandlingen og som klinisk indisert. Ved tiaminmangel skal behandlingen ikke startes før tiaminnivå er korrigert.Substitusjon av tiamin forhindrer utviklingen av Wernickes encefalopati og anbefales til alle som behandles med Fedratinib. Det er rapportert encefalopati ved bruk av fedratinib og tiamin analyse, evt oppstart substitusjon, ved oppstart er anbefalt. Kvalme kan også være et problem, spesielt ved starten.
Graviditet ved MPN
Myeloproliferative neoplasier øker risikoen for komplikasjoner i graviditeten, som tidlig abort, abruptio placenta, pre-eklampsi, og intrauterin vekstretardasjon. Abortrisikoen ved ET er 3- 4 ganger høyere enn i normalbefolkningen. Det er økt risiko for venøs trombose, særlig post partum, og risikoen er høyere ved tidligere trombose.
Det foreligger kun begrenset informasjon i litteraturen om behandlingen av disse sykdommene i graviditeten.
I nyere internasjonale retningslinjer skilles mellom høyrisiko graviditet (tidligere venøs eller arteriell trombose, tidligere MPN relatert blødning, tidligere MPN relatert svangerskapskomplikasjon slik som uforklart første trimester abort, intrauterin vekstretardasjon, intrauterin død eller dødfødsel, alvorlig pre-eklampsi, abruptio placenta, betydelig blødning pre- eller postpartum, og betydelig trombocytose (>1000-1500 x 109/L), og lavrisiko graviditet uten disse karakteristika.
Ved lavrisiko graviditet anbefales det at hematokrit holdes under 45 % (ved PV), bruk av lavdose acetylsalicylsyre 75 mg daglig fra graviditeten er konstatert, og profylaktisk lavmolekylært heparin i 6-8 uker etter partus. Acetylsalicylsyre bør seponeres 1 uke før forventet fødsel, erstattes med lavmolekylært heparin, og gjenopptas etter partus.
Ved høyrisiko graviditet anbefales det samme behandlingsopplegget. Ved tidligere trombose eller alvorlig svangerskapskomplikasjon bør man i tillegg vurdere lavmolekylært heparin profylakse dose gjennom hele graviditeten, men slutte med acetylsalicylsyre om det blir blødningskomplikasjoner. Hvis blodplatetallet blir høyere enn 1500 x 109/L, bør man vurdere interferon-a. Hvis det tidligere har forekommet alvorlig blødning, bør man unngå platehemmer og bruke interferon-alfa for å redusere trombocyttallet (Harrison et al., 2011). Interferon er kontraindisert ved amming, vesentlig pga manglende sikkerhetsdata.
Det er viktig at det er et nært samarbeid mellom hematolog og gynekolog ved behandling av gravide med MPN. Det er indisert med hyppig ultralyd inkludert doppler av arteria uterina for å kunne påvise vekstretardasjon eller redusert placentasirkulasjon, og derved behov for å oppgradere behandlingen.
For utfyllende opplysninger, se hovedprogrammet www.nmpn.org.
Trombotiske komplikasjoner ved MPN
MPN pasienter har betydelig forhøyet risiko for både tromboemboliske hendelser og blødning og dette er hovedårsaken til morbiditet og mortalitet ved MPN. Man bør ved alle former for trombeembolisk sykdom, både arterielle og venøse, tenke på bakenforliggende MPN. Særlig ved atypiske venetromboser (sinusvene trombose, Budd-Chiari og splanknikus venetromboser) kan MPN foreligge, også selv om de perifere hematologiske parametre er nærmest normale. MPN pasienter med JAK2 mutasjon er mest assosiert med trombose.
Det er derfor viktig å screene for og behandle risikofaktorer for vaskulære hendelser, slik som røking, hypertensjon, hyperlipidemi og diabetes mellitus hos pasienter med MPN.
Akutt arteriell trombose bør behandles som hos pasienter uten MPN i tillegg til rask reduksjon av hematokrit (ved PV) og forhøyet platetall. I alvorlige situasjoner (f.eks. cerebrale blødninger) kan flebotomi og plateaferese anvendes for å få rask reduksjon i celletall, men hydroxyurea bør startes så snart som mulig.
Akutt venøs trombose behandles også etter vanlige retningslinjer, og det er som ved arterielle tromboser viktig med normalisering av hematokrit og platetall, og HU bør startes så fort som mulig. Hos yngre kan man heller i rolig fase bytte til interferon-alfa.
Det er ingen prospektive studier som sammenligner alternative antikoagulasjonsbehandlinger ved MPN, og heller ingen studier som sammenligner korttids- og langtidsbehandling. Inntil prospektive studier foreligger, anbefaler de fleste warfarin hvis det er indikasjon for peroral antikoagulasjonsbehandling. Klinisk erfaring med bruk av DOAC i behandling og profylakse av trombose ser dog ut til å være trygt og blir mer og mer vanlig (Barbui et al., 2021). Kombinasjon av platehemming og antikoagulasjon er assosiert med ikke ubetydelig økt blødningsrisiko, og bør brukes med stor forsiktighet. Ved MPN og tidligere venøs trombose, og stor risiko for arteriell trombe kan kombinasjonen overveies.
Blødningskomplikasjoner ved MPN
Akvirert von Willebrands syndrom assosiert med høye platetall (over 1000 x109/L) er den vanligste årsaken til blødning ved ET og PV. Ved blødning og trombocytemi er viktigste tiltak å redusere platetallet. Hydroxyurea er det anbefalte medikamentet som gir raskest platefall. Interferon-alfa kan evt innsettes når man har god sykdomskontroll. Plateaferese kan være indisert ved ekstrem trombocytose hvor det er behov for meget rask reduksjon av platetallet, men må kombineres med cytoreduktiv behandling.
Kombinasjonen av Albyl E og anagrelid er assosiert med økt risiko for blødning, og bør brukes med forsiktighet.
Elektiv kirurgi
Pasienter med MPN har både økt risiko for trombose og blødning ved kirurgi. Generelt anbefales å bruke cytoreduktiv behandling for å normalisere blodverdiene før elektiv kirurgi. Etter større kirurgi må pasientene raskt restarte antikoagulasjon/platehemmer mtp trombotiske komplikasjoner, speselt abdominale etter splenektomi.
Kløe
Kløe kan være uutholdelig ved MPN og mange av pasientene har denne plagen ved diagnose. De fleste blir bedre etter oppstart venesectio og cytoreduktivbehandling. Antihistaminer eller serotonin reopptakshemmere (SSRI) kan også gi symptomlindring. Ruxolitinib har blitt rapportert å være effektiv hos pasienter med alvorlig kløe. Fototerapi med bruk av psoralen og ultrafiolett A lys har også vist å kunne hjelpe noen.
Kronisk lymfatisk leukemi (KLL)
Sist faglig oppdatert: 23.12.2021
Anbefalinger:
- Det er ikke indisert å behandle asymptomatiske pasienter i Binet stadium A (evidensgrad A).
- Det anbefales foreløpig ikke å velge behandling basert på risikofaktorer, fordi evidens for dette er mangelfull, med unntak av 17p-delesjon/TP53-mutasjon.
- Yngre (<65–70 år) pasienter i god form og uten vesentlig komorbiditet som har behandlingstrengende KLL bør behandles med FCR (fludarabin, syklofosfamid og rituksimab) når behandlingsmålet er livsforlengelse (evidensgrad A).
- Eldre (>65–70 år) pasienter og pasienter med betydelig komorbiditet bør også behandles med kjemoimmunterapi som førstelinjebehandling når behandlingsmålet er lengst mulig behandlingsfritt intervall (evidensgrad A). For de med god funksjonstatus er BR (bendamustine og rituksimab) det mest nærliggende alternativet. For de øvrige pasientene er klorambucil kombinert med anti-CD20 antistoff aktuelt (evidensgrad B).
- Det anbefales at behandlingen som ble gitt som førstelinjebehandling gjentas hos pasienter som residiverer etter initialt å ha respondert med lang (>12 mnd) progresjonsfri overlevelse og >36 mnd til ny behandlingsindikasjon. Anbefalingen baserer seg på klinisk erfaring og er ikke dokumentert ved kliniske studier (evidensgrad D).
- Pasienter som utvikler behandlingstrengende residiv før det er gått 12 måneder etter initial behandling med kjemoimmunterapi, kan behandles med signalveishemmerne ibrutinib eller idelalisib eller BCL-2 hemmeren venetoklaks, enten alene eller kombinert med anti-CD20 antistoff (evidensgrad A).
- Pasienter med fludarabin-resistent sykdom kan behandles med signalveishemmerne ibrutinib eller idelalisib eller BCL-2 hemmeren venetoklaks, enten alene eller med anti-CD20 antistoff (evidensgrad A).
- Følgende pasienter <70 år bør vurderes for allogen stamcelletransplantasjon:
- Noen av pasientene med del(17p)/TP53 mutasjon er potensielle kandidater for allogen stamcelletransplantasjon, men det er foreløpig ingen enkel måte å identifisere disse pasientene på før de oppviser svikt på behandling.
- Pasienter som svikter på behandling med signalveishemmere uavhengig av cytogenetisk eller molekylærgenetisk status kan være aktuelle transplantasjonskandidater.
- Det er en forutsetning for at godt behandlingsresultat ved allogen stamcelletransplantasjon at pasienten har liten restsykdom. Behandlingsstrategien fram til transplantasjon bør derfor ha som siktemål oppnå svært god sykdomskontroll hos de pasientene hvor allogen stamcelletransplantasjon vurderes som behandlingsalternativ.
- Autoimmune cytopenier behandles med Prednisolon 50 mg x 2 (eventuelt 1 mg/kg/d) i en uke, 25 mg x 3 i en uke og deretter avtrappende dosering alt etter effekt (evidensgrad C).
- Ved hyppige residiverende bakterielle infeksjoner (infeksjoner med kapselkledde bakterier) kan substitusjonsbehandling med immunglobulin være aktuelt hvis vaksinasjon (pneumokokkvaksine) ikke har ført fram og hypogammaglobulinemi forligger (evidensgrad B).
- Det er vanlig å anbefale pasienter med KLL influensavaksinasjon og vaksinasjon mot pneumokokker ved gjentatte pneumonier, men nytten er dårlig dokumentert (evidensgrad D).
- PCP-profylakse anbefales under og minst tre måneder etter behandling med fludarabinbaserte kombinasjonsregimer.
- Rituksimab eller rituksimab i kombinasjon med syklofosfamid er aktuelle alternativ ved AIHA, erytroaplasi og andre immunmedierte komplikasjoner som ikke responderer på prednisolon (evidensgrad C).
- R-CHOP er et vanlig brukt regime ved DLCBL (Richters transformasjon) (evidensgrad C). Dersom lymfomet ikke er klonalt relatert til KLL anbefales behandling som ved de novo DLBCL (evidensgrad D), mens det anbefales konsolidering med allogen stamcelletransplantasjon eller HMAS når lymfomet er klonalt relatert (evidensgrad D).
- R-CHOP kan også være et alternativ ved prolymfocytt leukemi i likhet med fludarabinbasert kjemoimmunoterapi (evidensgrad D).
Forekomst
Sist faglig oppdatert: 23.12.2021
Kronisk lymfatisk leukemi (KLL) er den hyppigste formen for leukemi i den vestlige verden og utgjør nær halvparten av alle tilfeller av leukemi hos pasienter over 65 år. Det er ca. 350 nye tilfeller av KLL i Norge hvert år. Median alder ved diagnosetidspunktet var i perioden 2003–2012 71,8 år i et norsk materiale (Ly et al., 1998). Om lag 45 % av pasientene var under 70 år, 18 % under 60 år og 26 % over 80 år ved diagnosetidspunktet. KLL ses hyppigere hos menn enn kvinner, og mann/kvinne ratio var 1,37 i perioden 2003–2012 som er uendret fra perioden 1953–1962 (Rousselot et al., 2014).
KLL utvikler seg fra en monoklonal B-lymfocytose som ofte er påvisbar mange år før sykdommen blir klinisk manifest (Landgren et al., 2009). Monoklonal B-lymfocytose kan påvises hos drøyt 3 % av antatt friske over 50 år i den vestlige verden, og transformasjonsraten til KLL er anslått til 1 % per år (Rawstron et al., 2008). Dette er helt parallelt til monoklonal gammopati og myelomatose både når det gjelder hyppighet og transformasjonsrate.
Det er ingen holdepunkter for at eksposisjon for kjemikalier, stråling, kosthold, røyking, virusinfeksjoner eller autoimmune sykdommer disponerer for KLL. Familiær disposisjon for lymfoproliferativ sykdom er den eneste veldokumenterte risikofaktoren for å få KLL med en relativ risiko på 6–8 når en førstegradsslektning har KLL (Tjønnfjord et al., 2012). KLL viser også store etniske forskjeller når det gjelder insidens, og det er særlig høy insidens i Skandinavia.
Diagnose og utredning
Sist faglig oppdatert: 23.12.2021
Sikker diagnose krever vedvarende lymfocytose >5,0 x 109/L i blod med karakteristisk immunfenotype dokumentert ved væskestrømscytometrisk undersøkelse eller tilsvarende (se avsnittene "Morfologi" og "Immunfenotyping", under). Ytterligere utredning er ikke nødvendig for å stille diagnosen KLL.
Klonal B-lymfocytose, hvor B-lymfocyttene utgjør <5 x 109/L hos ellers friske personer omtales som monoklonal B-lymfocytose. Småcellet lymfocyttært lymfom (SLL) defineres ved lymfeknutesvulst og/eller splenomegali og klonale B-lymfocytter i blod <5 x 109/L. Diagnosen bør verifiseres ved immunhistokjemisk undersøkelse av lymfeknutebiopsi så sant det er mulig.
Ovenstående diagnostiske kriterier er i tråd med de internasjonale anbefalingene om diagnostikk og behandling av KLL (Cheson et al., 1996; Hallek et al., 2008).
Morfologi
Leukemicellene ved KLL er små eller mellomstore lymfocytter med kondensert kjernekromatin (furubark-preg), ubetydelig eller ingen nukleolus og svært sparsomt cytoplasma (høy kjerne/cytoplasma ratio). Et stort antall kjerneskygger er svært karakteristisk for KLL (Gumprechtske kjerneskygger). Det finnes dog unntak fra morfologien som beskrevet over; for eksempel er lymfocyttene oftest ganske store og har lavere kjerne/cytoplasmaratio ved KLL med trisomi 12. Det er vanlig å finne et mindre antall litt større lymfocytter (<5 %) med lavere kjerne/cytoplasma ratio og tydelig nukleolus (prolymfocytter). Dersom disse cellene utgjør >55 % av lymfocyttene dreier det seg om prolymfocytt leukemi.
Immunfenotyping
Immunfenotyping bør som et minimum omfatte undersøkelse av CD5, CD19, CD20, CD23, CD22/CD79β, CD200, kappa og lambda slik at det kan utledes en KLL-skår.
Tabell 7.1: Immunfenotypisk KLL skår
Markør | Typisk KLL (skår) | Andre B-celle leukemier/lymfomer (skår) |
|---|---|---|
Membran Ig (kappa/lambda) | Svak (1) | Moderat/sterk (0) |
CD5 | Positiv (1) | Negativ (0) |
CD23 | Positiv (1) | Negativ (0) |
CD79β eller CD22 | Svak/negativ (1) | Moderat/sterk (0) |
CD200 | Positiv (1) | Negativ (0) |
Skår vil være 4–5 i ca. 90 % av tilfellene med KLL og 0–1 i en tilsvarende andel andre primære B-celle leukemier/lymfomer. Henholdsvis 6 % og 2 % av pasientene med KLL har skår 3 og 1–2.
Klinisk undersøkelse
En fullstendig klinisk undersøkelse med vekt på om det foreligger lokalisert eller generell lymfeknutesvulst og hepatosplenomegali er obligatorisk. Undersøkelsen bør også dokumentere høyde og vekt.
Anamnesen må avklare om det er dreier seg om asymptomatisk eller symptomatisk KLL og om det foreligger B-symptomer. Hastigheten i sykdomsutviklingen bør forsøkes avdekket ved å innhente tidligere blodprøveresultat hvis slike finnes. Det bør gjøres en grundig infeksjonsanamnese, og forekomst av autoimmune sykdommer bør avdekkes. Anamnesen bør også forsøke å avdekke familiær forekomst av lymfoproliferativ sykdom eller annen hematologisk malignitet.
Anamnesen må avklare om pasienten har komorbiditet som eventuelt kan få betydning for valg av behandlingsmål og behandlingsstrategi.
Andre undersøkelser
Beinmargsaspirat og/eller biopsi kreves ikke for å stille diagnosen.
Beinmargsundersøkelse kan være nyttig ved cytopenier for å avklare årsakene til cytopenien(e), og undersøkelsen vil dermed være veiledende for behandling. Beinmargsundersøkelse og/eller lymfeknutebiopsi kan bidra med diagnostisk informasjon ved atypisk lymfocyttmorfologi, lav KLL-skår (≤3) eller ved mistanke om SLL.
CT eller ultralydundersøkelser er ikke en nødvendig del av den diagnostiske utredningen ved KLL, og slike undersøkelser bør ikke gjøres annet enn på spesifikk indikasjon.
Andre undersøkelser som kan være nyttige for vurdering av pasienten, men ikke nødvendige for diagnosen:
- Full blodcelletelling (inkluderer differensialtelling, reticulocytter og erytrocyttindekser)
- Proteinelektroforese i serum/plasma, kvantitering av Ig og β2-mikroglobulin
- Haptoglobin og direkte antiglobulin test (DAT)
- Kreatinin og urinsyre
- Bilirubin, LD og transaminaser
Stadieinndeling
Stadieinndeling etter Binet eller Rai (Binet et al., 1981; Rai et al., 1975) er en enkel måte å beskrive tumorvolum og prognose ved KLL (tabell 7.2). Stadium er basert på en kombinasjon av klinisk vurdering av tumorvolum (lymfeknutesvulst og organomegali bedømmes palpatorisk) og graden av leukemisk beinmargaffeksjon (uttrykt som anemi og/eller trombocytopeni). Binets system brukes i Norge og Europa.
Tabell 7.2: Stadieinndeling av KLL etter Binet
Stadium | A | B | C |
|---|---|---|---|
Antall involverte lymfeknuteregioner | 0–2 | 3–5 | 0–5 |
Hemoglobin (g/dL) | >10 | >10 | <10 |
Trombocytter (109/L) | >100 | >100 | <100 |
5 definerte lymfoide regioner: hals (inkludere lymfoide organer i nese-svelgrommet), aksiller, lysker, milt og lever. Lymfeknutesvulst i en region gis vekttall 1 uavhengig av om affeksjonen er ensidig eller dobbeltsidig.
Vi anbefaler at sykdomsstadium eksplisitt beskrives ved diagnose, ved tegn til sykdomsprogresjon og hver gang en ny behandlingsserie startes (konferer behandlingsindikasjoner og responsevaluering). Kun anemi og/eller trombocytopeni som skyldes leukemisk beinmargsaffeksjon inngår i stadieinndelingen. Cytopenier som er immunmedierte som autoimmun hemolytisk anemi, immunmediert trombocytopeni eller immunmediert erytroaplasi teller ikke med. Det kan være vanskelig å avgjøre om cytopenien(e) er sekundær til massiv beinmargsaffeksjon eller er immunmediert. Beinmargsundersøkelse bør gjøres som ledd i avklaringen.
Differentialdiagnoser
I de aller fleste tilfeller vil vedvarende leukocytose forårsaket av små modne lymfocytter vise seg å være KLL. Nøye vurdering av blodutstryk vil avsløre de fleste tilfeller hvor lymfocytosen skyldes annen sykdom, og i disse tilfellene vil endelig diagnose nesten alltid kunne sikres ved væskestrømscytometrisk immunfenotyping og/eller cytogenetisk/molekylærgenetisk undersøkelse av lymfocytter i blod (Müller-Hermelink et al., 2001).
CD5+CD19/20+ kronisk lymfoproliferativ sykdom
Mantelcelle lymfom med leukemisering
Marginal sone lymfom med leukemisering, spesielt den spleniske varianten
CD5-CD19/20+ kronisk lymfoproliferativ sykdom
B-prolymfocytt leukemi
Follikulært lymfom med leukemisering
Lymfoplasmacytisk lymfom med leukemisering
Hårcelle leukemi
Kronisk lymfoproliferativ sykdom av T-celle type
T-prolymfocytt leukemi
Storcellet granulær lymfocytt leukemi (NK- eller T-celle type)
Perifert T-celle lymfom med leukemisering
Prognose
Stadieinndeling
Sykdomsstadium har veldokumentert prognostisk betydning, og stadieinndeling etter Binet eller Rai separerer pasientene i tre grupper med forskjellig prognose (tabell 7.2). Som et resultat av langt mer effektiv behandling, har overlevelse for pasienter med behandlingstrengende sykdom blitt vesentlig bedre, slik at stadieinndelingen som et prognostisk instrument har fått mindre betydning. Stadieinndelingen har fortsatt stor betydning for gruppering av pasienter i kliniske studier.
Alminnelig tilgjengelige undersøkelser
β2-mikroglobulin er en rutineanalyse ved de fleste laboratorier for medisinsk biokjemi, og høye verdier av β2-mikroglobulin (>3 mg/L) er forbundet med langt dårligere prognose enn lave verdier. Et nomogram basert på β2-mikroglobulin og enkle kliniske parametre skiller godt mellom prognostiske grupper (Wierda et al., 2007). Pasienter som er DAT+ (uavhengig om de har hemolytisk anemi) har dårligere prognose enn de som er DAT- (Dearden et al., 2008).
Lymfocytt-doblingstid er også en enkel biologisk markør som har prognostisk verdi. Tradisjonelt har man benyttet lymfocytt-doblingstid kortere eller lengre enn 12 måneder som «cut-off».
Cytogenetiske avvik
Ved diagnose har ca. 70 % av alle KLL-pasienter cytogenetiske avvik ved FISH-analyser (fluorescens in situ hybridisering), og ca. 30 % har normal karyotype (Tjønnfjord et al., 2012).
Det er ingen av de cytogenetisk avvikene vi kjenner til ved KLL som er sykdomsspesifikke, og de cytogenetiske avvikene ved KLL oppfattes ikke som primære, men som sekundære avvik. De cytogenetiske avvikene er oftest, men ikke alltid, karakterisert ved tap av genetisk materiale, dvs. delesjoner (Döhner et al., 2000; Tjønnfjord et al., 2012).
Delesjon av den lange armen på kromosom 13, del(13q) er forbundet med god prognose. Trisomi av kromosom 12 er forbundet med intermediær prognose.
Delesjon av den lange armen på kromosom 11, del(11q) medfører ATM-tap/dysfunksjon og er relatert til relativt dårlig prognose.
Delesjon av den lange armen av kromosom 6, del(6q) er forbundet med framtredende lymfeknutesvulst og har intermediær prognose.
Delesjon av den korte armen av kromosom 17, del(17p) som involverer TP53-genet er vanligvis forbundet med defekt p 53-signalvei, dårlig respons på kjemo(immun)-terapi og kort overlevelse. KLL med p 53-dysfunksjon svarer vanligvis langt bedre på behandling med signalveishemmere eller BCL-2 hemmer, men avviket er til tross for det forbundet med dårlig prognose.
Normal karyotype er forbundet med relativt god prognose, men ikke fullt så god som hos pasienter med del(13q) som eneste cytogenetiske avvik.
Cytogenetiske avvik tilkommer gjerne under sykdomsforløpet som uttrykk for klonal evolusjon.
Molekylærgenetiske avvik
Del(17p) forekommer vanligvis i heterozygot form, men i ca. 75 % av tilfellene foreligger samtidig en mutasjon i det bevarte TP53-allelet og samlet gir dette opphav til p 53-dysfunksjon. Ca halvparten av de KLL pasientene som har TP53 mutasjon har ikke del17p.TP53-mutasjonsanalyse er rutinemessig tilgjengelig i Norge.
Den cytotoksiske effekten av de fleste konvensjonelle medikamentene som benyttes ved behandling av KLL er avhengig av en fungerende p 53-signalvei.
IGHV-genets mutasjonstatus
Somatisk hypermutasjon i KLL-cellenes foretrukne IGHV-gen har betydning for prognosen (Damle et al., 1999; Hamblin et al., 1999). KLL med umutert IGHV-gen (≥98 % homologi med kimbanen) er forbundet med vesentlig dårligere prognose enn KLL med mutert IGHV-gen (≤98 % homologi med kimbanen); median overlevelse henholdsvis 95 mnd og 293 mnd (basert på data før kjemoimmunterapi ble standard behandling).
Foretrukket IGHV-gen er også av betydning. KLL hvor leukemicellene benytter IGHV-genet 3–21 er forbundet med dårlig prognose, og prognosen er i dette tilfellet uavhengig av om IGHV-genet 3–21 er mutert eller ikke (Tobin et al., 2002).
Immunfenotypiske karakteristika
Umutert IGHV-gen er ofte assosiert med høy ekspresjon av overflatemolekylet CD38 (Damle et al., 1999). Umutert IGHV-gen er også assosiert med høy ekspresjon av intracytoplasmatisk ZAP-70 (ζ-assosiert protein 70) (Crespo et al., 2003). Høy ekspresjon av CD38 og/eller ZAP-70 har uavhengig prognostisk betydning, men ingen av disse immunfenotypiske markørene er fullgode surrogatmarkører for IGHV-genets mutasjonsstatus. CD38 ekspresjon angis av flere laboratorier ved besvarelse av immunfenotyping ved KLL. Metoden for bestemmelse ZAP-70 ekspresjon er ikke tilfredsstillende standardisert og brukes ikke i rutinediagnostikken.
Bruk av prognostiske markører
Nærmere 90 % av pasientene presenterer seg i et tidlig sykdomsstadium, og de er vanligvis asymptomatiske og ikke behandlingstrengende. For mange pasienter vil det være av betydning å få informasjon om sannsynligheten for rask progresjon og snarlig behandlingsbehov eller om de kan forvente et mer indolent sykdomsforløp som vil være forløpet hos de fleste pasientene. IGHV-mutasjonsstatus og CD38 ekspresjon ved diagnosetidspunkt har best prognostisk utsagnskraft i denne situasjonen (Pepper et al., 2012). Ikke-publiserte internasjonale erfaringer tilsier at ca. 75 % av pasienter med mutert KLL aldri trenger behandling, og ca. 25 % av pasientene med umutert KLL trenger heller aldri behandling. FISH-analyse og TP53-mutasjonsanalyse har også prognostisk utsagnskraft, men cytogenetiske avvik kan tilkomme under tiden. Disse undersøkelsen bør derfor bare gjøres ved sykdomsprogresjon og behov for behandling for å avdekke pasientene som har KLL karakterisert ved del(17p) og/eller TP53-mutasjon, og som bør vurderes for behandling med signalveishemmere eller BCL-2 hemmer.
Behandling
Sist faglig oppdatert: 21.12.2023
Asymptomatiske pasienter
Ved diagnose er de fleste (85–90 %) pasientene i Binet stadium A, og diagnosen er oftest stilt ved en tilfeldighet. Tidlig behandling med kjemoterapi fører ikke til bedret overlevelse (Pepper et al., 2012), og vi venter fortsatt på resultatet av kliniske studier med de nye behandlignsalternativene. Standard behandling ved tidlig sykdomsstadium er derfor stadig «watch-and-wait». Det er konsensus om at småcellet lymfocytært lymfom (SLL) handteres på samme måte som KLL.
- Det er ikke indisert å behandle asymptomatiske pasienter i Binet stadium A (evidensgrad A).
Indikasjon for behandling
The National Cancer Institute (NCI) utarbeidet i 1996 en anbefaling om behandlingsindikasjoner ved KLL (CLL Trialists' Collaborative Group, 1999), og disse anbefalingene har fortsatt gyldighet (Hallek et al., 2008). De fleste pasientene i Binet stadium B og C og noen pasienter i Binet stadium A med progressiv sykdom er behandlingstrengende etter disse internasjonalt anerkjente kriteriene (tabell 7.3). Det har vært iverksatt studier for å avdekke om høyrisikopasienter i Binet stadium A er tjent med behandling, men ingen resultater fra disse studiene er så langt publisert.
Tabell 7.3: Indikasjoner for behandling
- Progressiv beinmargssvikt
- Utvikling av eller forverrelse av anemi
- Utvikling av eller forverrelse av trombocytopeni
- Hb <10 g/dL eller trombocytter <100x109/L ses vanligvis som behandlingsindikasjon, men trombocytter <100x109/L kan holde seg stabile over en lang periode og krever ikke
nødvendigvis umiddelbar behandling
- Massiv (>10 cm) eller progressiv lymfeknutesvulst
- Massiv (>6 cm) eller progressiv splenomegali
- Progressiv lymfocytose
- >50 % økning i løpet av 2 måneder
- Lymfocytt-doblingstid <6 måneder*
- Allmenn symptomer
- Vekttap (>10 %) i løpet av siste 6 måneder
- Feber >38 °C i mer enn 2 uker
- Uttalt fatique; ikke i stand til å arbeide eller utføre normale aktiviteter
- Nattesvette
- Autoimmune cytopenier resistente for steroidbehandling
* Lymfocytt-doblingstid bør vurderes på basis av absolutt antall lymfocytter (ALC) målt med to ukers intervall sammenlignet med to observasjoner med to ukers mellomrom etter 3 måneder.
Før behandlingsstart bør det utarbeides en individuell behandlingsplan på basis av relevante risikofaktorer, komorbiditet og prognostiske markører. Behandlingsmålet bør være klart formulert i forståelse med pasienten, og en plan for hvordan man vil vurdere behandlingsrespons bør være lagt (se tabell 7.4).
Tabell 7.4: Responskriterier til bruk i klinisk praksis (Tjønnfjord et al., 2012)
Kriterium | Komplett remisjon, KR | Partiell remisjon, PR | Progressiv sykdom, PS |
|---|---|---|---|
Symptomer | Ingen | Ingen |
|
Lymfeknuter | Ingen lymfeknuter med diameter >1.5 cm | >50 % reduksjon | >50 % økning eller nye manifestasjoner |
Lever/milt | Ikke palpabel | >50 % reduksjon | >50 % økning eller nye manifestasjoner |
Hemoglobin | >11 g/dL | >11 g/dL eller 50 % bedring |
|
Granulocytter | >1.5 x 109/L | >1.5 x 109/L eller 50 % bedring |
|
Lymfocytter | <4 x 109/L | >50 % reduksjon | >50 % økning |
Trombocytter | >100 x 109/L | >100 x 109/L eller 50 % bedring |
|
BM-aspirat | <30 % lymfocytter | BM aspirat ikke relevant |
|
BM-biopsi | Ingen nodulære KLL-infiltrater i en normo- eller hypocellulær beinmarg | BM biopsi ikke relevant |
|
Tabell 7.5: Responskriterier til bruk dersom behandlingsmålet er livsforlengelse
(Tjønnfjord et al., 2012)
CR | PR |
|---|---|
Ingen klonal lymfocytose i blod | >50 % reduksjon av lymfocytose |
Ingen lymfeknutesvulst | >50 % reduksjon av lymfeknutesvulst |
Ingen hepatosplenomegali (klinisk) | >50 % reduksjon av hepatosplenomegali |
Ingen B-symptomer | Minst en av følgende: |
Nøytrofile granulocytter >1,5x109/L | Nøytrofile granulocytter >1,5x109/L |
Tromboytter >100x109/Lβ2 | Trombocytter >100x109/L |
Hemoglobin >11,0 g/dL | Hemoglobin >11,0 g/dL |
BM uten klonale lymfocytter | eller minst 50 % bedring fra utgangsverdi |
CRi |
|
Som CR, men ikke hematologisk rekonstitusjon |
|
Undersøkelser før behandlingsstart
Klinisk vurdering og undersøkelse som gir grunnlag for stadieinndeling, funksjonsnivå (ECOG eller WHO) og komorbiditet. Objektiv angivelse av lymfeknutestørrelse og lever- og miltstørrelse i cm basert på klinisk undersøkelse er viktig for å kunne vurdere behandlingseffekt.
Full blodcelletelling med differensialtelling og reticulocytter
Vurdering av blodutstryk
Proteinelektroforese i serum/plasma, kvantitering av Ig og β2-mikroglobulin
Haptoglobin og direkte antiglobulin test (DAT)
Kreatinin og urinsyre
Bilirubin, LD og transaminaser
Beinmargsundersøkelse (aspirat og biopsi) anses ikke obligatorisk før behandlingsstart, men undersøkelsen kan bidra med viktig informasjon som veiledning for behandlingen og anbefales hos pasienter hvor behandlingsmålet er komplett remisjon.
CT- og ultralydundersøkelser har liten verdi i behandling og oppfølging av pasienter med KLL (Eichhorst et al., 2011), og disse undersøkelsene bør kun gjøres på spesifikk indikasjon.
Infeksjonsstatus med rtg. thorax og serologiske undersøkelser med tanke på HIV, HCV, HBV, EBV og CMV bør utføres hos alle pasienter hvor det er aktuelt med behandling pga risiko for virus-reaktivering.
Hvis det er grunn til å mistenke transformasjon (isolert stor lymfeknutesvulst, allmennsymptomer, høy LD etc) bør det gjøres biopsi for å avklare om det foreligger transformasjon. PET-undersøkelse kan være til hjelp for å identifisere de(n) lymfeknuten(e) som er best egnet for biopsi hvis dette ikke er åpenbart ved klinisk undersøkelse.
Tabell 7.6: Undersøkelser før behandlingsstart og ved responsevaluering i klinisk praksis
Undersøkelse | Før behandling | Ved responsevaluering |
|---|---|---|
Sykehistorie/funksjonsnivå | Alltid | Alltid |
Klinisk undersøkelse | Alltid | Alltid |
Komplett blodcelletall | Alltid | Alltid |
Beinmargsundersøkelse | Ønskelig | Alltid |
Biokjemi (lever/nyre) | Alltid | Alltid |
Immunglobuliner | Ønskelig |
|
Direkte antiglobulintest (DAT) | Alltid | Alltid |
HIV, HCV, HBV, CMV, EBV | Alltid |
|
Rtg. thorax | Alltid |
|
Cytogenetikk (FISH) | Alltid |
|
TP53 mutasjonsanalyse | Alltid |
|
IGHV mutasjonsstatus | Alltid, viss ikke allerede gjort |
|
Bildediagnostikk; CT og/eller ultralydundersøkelser | På spesifikk indikasjon | På spesifikk indikasjon |
Førstelinjebehandling
- Det anbefales å velge behandling basert på sykdomsstadium, funksjonsnivå, komorbiditet og prognostiske faktorer som IGHV-mutasjonsstatus og genetiske avvik, spesielt del(17p)/TP53-mutasjon.
Det er følgende førstelinjebehandlinger tilgjengelig:
- Tidsbegrenset kjemoimmunterapi (fludarabin og sykolfosfamid, bendamustine eller klorambucil i kombinasjon med et anti-CD20 antistoff)
- Kontinuerlig behandling med Bruton kinase hemmer(e) (BTKis) eller fosfatidylinositol-3 kinase p 110δ (PI3Kδ) hemmer inntil progresjon eller bivirkninger som nødvendiggjør seponering
- Tidsbegrenset behandling med BCL-2 hemmer i kombinasjon med et anti-CD20 antistoff
Ved valg av behandling skal IGHV-mutasjonsstatus og del(17p)/TP53-mutasjon status legges til grunn og dessuten pasient-relaterte faktorer som ko-medikasjon, komorbiditet, pasientens preferanser, tilgang på medikamenter og pasientens even til å gjennomføre behandling som planlagt.
CLL8-studien var den første randomiserte studien med god design som viste gevinst av en førstelinjebehandling på totaloverlevelse ved KLL (Hallek et al., 2010). FCR-regimet (fludarabin, syklofosfamid, rituximab) bedret totaloverlevelsen etter tre år med 5 % sammenliknet med FC. Overlevelsesgevinsten var begrenset til pasienter i Binet stadium A og B og til pasienter under 65 år, mens pasienter i Binet stadium C ikke hadde noen sikker overlevelsesgevinst. HOVON 68 CLL-studien som sammenlignet FC med FC+alemtuzumab kom fram til lignende resultater (Hallek et al., 2010).
Flere observasjonsstudier og en nylig publisert meta-analyse viser at det er KLL-pasienter med mutert IGHV-gen som har spesielt god nytte av FCR-behandling. Mange pasienter når komplett remisjon med MRD-negativitet, og disse pasientene har svært langvarig sykdomsfri overlevelse. Overlevelseskurvene indikerer at et platå nås hos disse pasientene som for praktiske formål blir å betrakte som kurerte.
Behandling med ibrutinib inntil progresjon enten som monoterapi eller i kombinasjon med anti-CD20 antistoff har resultert i lengre progresjonsfri overlevelse (PFS) enn kjemoimmunterapi (fludarabin og syklofosfamid, bendamustin eller klorambucil i kombinasjon med anti-CD20 antistoff) (Byrd et al., 2013; Hillmen et al., 2007; Pettitt et al., 2009). Det er nå rimelig godt dokumentert at anti-CD20 antistoff ikke gir noen ekstra behandlingsgevinst i kombinasjon med ibrutinib (Furman et al., 2014). Derimot er det ikke klarlagt hva som er optimal behandlingslengde ved behandling med ibrutinib.
En nylig publisert fase III studie som sammenlignet ibrutinib i kombinasjon med rituksimab med FCR hos yngre pasienter i god form antyder at det er en overlevelsesgevisnt (OS) hos pasientene som ble behandlet med ibrutinib (Hillmen et al., 2007), mens to tilsvarende studier som tillot cross-over for pasienter med progresjon etter kjemoimmunterapi ikke viste bedret OS. Men disse studiene hvor kjemoimmunbehandling var henholdsvis bendamustin + rituksimab og klorambucil + obinutuzumab viste at behandling med ibrutinib ga en betydelig og statistisk signifikant bedret PFS (Byrd et al., 2013; Pettitt et al., 2009). Dette var pasienter som ikke ble funnet egnet for FCR pga. alder, funskjosnnivå og komorbiditet. Tilsvarende forskjeller så man også i en studie der 2. generasjons BTKi’en acalabrutinib med og uten obinutuzumab ble sammen lignet med klorambucil + obinutuzumab. Subgruppe-analyse fra disse studiene viser at det særlig er pasientene med umutert KLL som har utbytte av behandling med BTKis.
Studier med BCL-2 hemmeren venetoklaks i kombinasjon med obinutuzumab (tidsbegrenset) sammenlignet med bendamustin + rituksimab hos komorbide pasienter har også vist betydelig gevinst når det gjelder PFS for pasientene som ble behandlet med venetoklaks + rituksimab (Eichhorst et al., 2009). Det er tilsvarende resultater fra en studie som har sammenlignet klorambucil + obinutuzumab og venetoklaks + obinutuzumab (Catovsky et al., 2007). Det er ikke gjort studier med venetoklaks i sammenligning med FCR hos yngre pasienter, og det er heller ikke gjort studier for å avklare kombinasjonsbehandling med anti-CD20 antistoff gir en tilleggsgevinst sammenlignet med venetoklaks monoterapi.
Det er foreløpig ikke publisert studier som sammenligner venetoklaks-basert behandling med BTKi-basert behandling, så valget mellom det ene eller andre behandlingsregimet må basere seg på forhold som tidsbegrenset behandling, bivirkningsprofil, administrasjonsmåte for medikasjonen, kontrollintensieten og kontrollengden.
Pasienter med KLL med TP53-mutasjon eller del(17p) bør ha behandling med BTKis. Kjemoimmunterapi er ikke en behandlingsopsjon pga. dårlig prognose ved kjemoimmunterapi uavhengig av IGHV mutasjonsstatus. BCL-2 hemmeren venetoklaks er et alternativ til BTKIs hos disse pasientene. Kardiovaskulær sykdom, samtidig antikoagulasjonsbehandling, samtidig behandling med platehemmere og/eller behandling med sterke CYP3A4-hemmere er av betydning for valget mellom dise to behandlingsalternativene. Enn så lenge er erfaringen større med venetoklaks som behandling ved svikt på behandling med BTKis enn erfaringen med BTKis ved svikt av venetoklaks. Fosfatidylinositol 3 kinase (PI3K) hemmeren idelalisib er et alterantiv når verken BTKis eller BCL-2 hemmer kan benyttes.
- Yngre (<65–70 år) pasienter i god form og uten vesentlig komorbiditet som har behandlingstrengende mutert KLL bør behandles med FCR (fludarabin, syklofosfamid og rituximab) når behandlingsmålet er livsforlengelse (evidensgrad A).
- Yngre (<65–70 år) pasienter i god form og uten vesentlig komorbiditet som har behandlingstrengende umutert KLL bør behandles med ibrutinib, men inntil det foreligger et vedtak hos Beslutningsforum som gir åpning for slik behandling er FCR behandlingsvalget (evidensgrad A).
- Eldre (>65–70 år) med god funksjonstatus og mutert KLL; BR (bendamustin og rituksimab) er et godt behandlingsalternativ (evidensgrad A).
- Eldre (>65–70 år) pasienter og pasienter med betydelig komorbiditet bør behandles med behandles med BTKIs eller BCL-2 hemmer, men inntil det foreligger et vedtak hos Beslutningsforum som gir åpning for slik behandling er kjemoimmunterapi (klorambucil + anti-CD20 antistoff) behandlingsvalget (evidensgrad A).
- Pasienter med del(17p)TP53-mutasjon og behandlingsindikasjon bør vurderes for behandling med BTKIs eller BCL-2 hemmer uavhengig av alder (evidensgrad A).
Basert utelukkende på medisinske vurderinger synes kjemoimmunterapi (FCR) bare å være indisert som førstelinjebehandling hos yngre pasienter uten komorbiditet og mutert KLL. Mens det ved de fleste andre situasjoner er en gevinst å velge enn annen førstelinjebehandling enn kjemoimmunterapi. Kostnader utforderer en slik anbefaling. En nylig analyse konkludert med at det må en ganske betydelig prisreduksjon til for at førstelinjebehandling med BTKIs skal være kostnadseffektiv førstelinjebehandling (Goede et al., 2014). Vurdering i Nye Metoder mai 2021 konkluerte med at Venetoklaks kan benyttes i kombinasjon med et CD20-antistoff i førstelinjebehandling hos pasienter med 17p-delesjon, TP53 mutasjon eller 11q-delesjon.
Andrelinjebehandling og seinere behandling
Ved residiv bør behandling først startes når pasienten har symptomer. Mange pasienter med residiv kan følges i en lengre periode uten å trenge behandling. Sjøl stopp av BTKis eller venetoklaks pga. bivirkninger medfører ikke et umiddelbart behov for alternativ behandling. Saken stiller seg naturligvis annerledes om behandlingen med BTKIs eller venetoklaks stoppes pga. progresjon.
Før valg av andrelinje og seinere behandling bør man få oversikt over tidligere gitt behandling, repons på tidligere behandling og remisjonsvarighet. Unngå å gjenta tidligere behandling dersom behandlingen ikke har gitt tilfredstillende respons. Cytogenetisk undersøkelse (karyotypering/FISH) med tanke på del(17p) og molekylærgenetisk undersøkelse med tanke på TP53-mutasjon bør gjøres før hver ny behandlingslinje. Andelen pasienter som del(17p) og/eller TP53-mutasjon øker for hver behandlingslinje, og disse pasientene trenger annen behandling enn kjeomimmunterapi. Behandlingsmulighetene er:
- Venetoklaks + anti-CD20 antistoff inntil KR, MRD negativ eller inntil 24 måneder
- Kontinuerlig behandling med BTKIs (per i dag er kun ibrutinib aktuell) eller PI3KI
Etter vurdering Nye Metoder 2023 ble det besluttet at venetoklaks (Venclyxto) ikke kan brukes i kombinasjon med rituksimab (VR) til behandling av voksne pasienter med kronisk lymfatisk leukemi (KLL) som tidligere har mottatt minst en behandling med BTK eller BCL-2-hemmer.
Fornyet behandling med kjemoimmunterapi hos pasienter hvor det ikke er påvist del(17p)/TP53-mutasjon er bare aktuelt hvis behandlingsrepsonsen har vart minst 3 år. Det bør utvises forsiktighet ved rebehandling med FCR pga. økt risiko for toksisitet og sekundær myeloid malignitet (evidensgrad B)
- Ved symptomatisk residiv innen 3 år etter tidsbegrenset behandling eller non-respons på behandling bør behandlingsregimet endres uavhengig av førstelinje behandling (evidensgrad A).
- Ved symptomatisk residiv mer enn 3 år etter tidsbegrenset behandling kan det være aktuelt å gjenta primærbehandling gitt at fornyet utredning ikke har avdekket del(17p) eller TP53-mutasjon (evidensgrad B).
- Pasienter som utvikler behandlingstrengende residiv før det er gått 3 år etter initial behandling med kjemoterapi bør behandles med BTKIs, PI3KI eller venetoklaks eventuelt med tillegg av anti-CD20 antistoff (evidensgrad A).
- Yngre pasienter som responderer på behandlingen bør vurderes for allogen stamcelletransplantasjon ved alder <70 år dersom de hadde primært resistent sykdom eller kort tid (<12 måneder) til andrelinje behandling (evidensgrad B).
Allogen stamcelletransplantasjon
Allogen stamcelletransplantasjon anses som etablert behandling hos utvalgte pasienter med KLL, og man kan forvente 40–50 % 5 års sykdomsfri overlevelse og 60–65 % 5 års totaloverlevelse hos pasienter transplantert pga. KLL med svært dårlig prognose (Dreger et al., 2010). Behandlingsresultatene, som alle er fra ikke-randomiserte studier, viser at progresjonsfri overlevelse og total overlevelse ikke skiller seg særlig mye fra hverandre om transplantasjonen etterfølger myeloablativ kondisjonering eller doseredusert kondisjonering, men årsaken til behandlingssvikt er ulik.
Den europeiske blod- og beinmargstransplantasjonsgruppen (EBMT) utarbeidet konsensusretningslinjer for allogen stamcelletransplantasjon (Dreger et al., 2007), og disse ble adaptert av det norske transplantasjonsmiljøet. Disse retningslinjene var basert på de behandlingsalgoritmene vi hadde før introduksjonen av BTKIs og BCL-2 hemmere. Nå er svært mange av den oppfatning at allogen stamcelletransplantasjon har en mer beskjeden plass ved KLL. Det knytter seg stor usikkerhet til hvordan vi skal identifisere de få pasientene som er kandiater for allogen stamcelletransplantasjon.
- Følgende pasienter <70 år kan vurderes for allogen stamcelletransplantasjon:
- Pasienter med del(17p)/TP53-mutasjon med tegn til behandlingssvikt på førstelinjebehandling med signalveishemmer (evidensgrad B).
- Pasienter i seinere behandlingslinjer med tegn til behandlingssvikt på behandling med signalveishemmere uavhengig av del(17p)- eller TP53-mutasjonsstatus (evidensgrad B).
- Pasienter med klonalt relatert Richters transformasjon og som kan bringes i remisjon med R-CHOP eller lignende behandling (evidensgrad D).
De viktigste grunnene til dårlig behandlingsresultat ved allogen stamcelletransplantasjon er manglende sykdomskontroll ved transplantasjonstidspunktet. Det er derfor viktig at potensielle transplantasjonskandidater identifiseres på et tidlig tidspunkt og at transplantasjonen gjennomføres mens sykdommen fortsatt er behandlingsfølsom. Nødvendig cytoreduktiv behandling kan da gjennomføres i forkant av transplantasjonen.
Strålebehandling
KLL er en systemisk sykdom, og kjemoimmunterapi er den viktigste behandlingsmodaliteten. Lymfoproliferative sykdommer er imidlertid svært følsomme for strålebehandling.
Miltbestråling tolereres vanligvis godt, og behandlingen gir god symptomatisk effekt hos 50–90 % av pasientene (Guiney et al., 1989). Miltbestråling er svært lite i bruk.
Strålebehandling gir også oftest effektiv palliasjon hos pasienter med plagsomme «bulky» lymfeknutemasser, og det kan være tilstrekkelig med lavere doser enn hva som konvensjonelt gis for å kontrollere lokale tumor masser (Girinsky et al., 2001).
Splenektomi
Splenektomi har aldri vært sammenlignet i randomiserte studier med annen behandling ved KLL. Dersom indikasjonen er anemi eller trombocytopeni kan en forvente effekt hos henholdsvis 50–77 % og 61–88 % av pasientene (Cusack et al., 1997; Seymour et al., 1997).
De aksepterte indikasjonene for splenektomi er symtomgivende massiv splenomegali og refraktære cytopenier.
Behandlingsregimer
Klorambucil var standard førstelinjebehandling ved KLL i mange år. Responsraten er doseavhengig og har vært rapportert mellom 45–86 %. Tillegg av prednisolon til klorambucil gir ingen gevinst på overlevelsen og anbefales kun på indikasjonen samtidig autoimmun sykdom. Tillegg av rituksimab i doser som angitt under FCR eller obinutuzumab øker responsraten, responsvarigheten og totaloverlevelsen.
- Intermitterende klorambucil: 15 mg/m2 p.o. i 4 dager hver 28. dag til maksimalrespons er mest brukt.
- Kontinuerlig klorambucil: 3 mg/m2 p.o daglig (ca. 6 mg/dag som startdose) justert slik at det oppnås moderat neutropeni/trombopeni, brukes noen steder. Gir mindre kvalme enn intermitterende behandling.
Det er viktig at det gis tilstrekkelig høy dose med klorambucil, og at dosen justeres etter den toksiske effekten på beinmargen. Ta prøver etter ca. 2 uker ved intermitterende behandling. Reduser dosen med 25 % ved granulocytopeni/trombocytopeni (granulocytter <0,5 x 109/L og/eller trombocytter <50 x 109/L, grad 3 toksisitet) ved nadir. Øk dosen med 25 % hvis det ikke kommer fall i granulocytter/trombocytter ved nadir.
Behandling fortsettes til det ikke lenger kommer bedring av responsen vurdert etter objektive responskriterier (tabell 7.5) og seponeres deretter. 4–6 måneders behandling er vanlig. Fortsatt behandling når maksimal respons er oppnådd utsetter pasienten for bivirkninger og bør unngås.
Bendamustin brukes både som førstelinjebehandling og som annenlinjebehandling.
- Bendamustin: 90 mg/m2 iv dag 1 og 2 som førstelinjebehandling.
Ved kombinasjon med rituksimab gis 90 mg/ m2 dag 1 og 2.
- Bendamustin 70 mg/m2 dag 1 og 2 ved residivbehandling, med ny kur hver 28. dag. Dosereduksjon ved nøytropeni/trombopeni, se Felleskatalogen. Det er ikke nødvendig med dosereduksjon ved nyresvikt.
Fludarabin og syklofosfamid (FC) gir høyere responsrate og lengre progresjonsfri overlevelse enn fludarabin monoterapi. Fludarabin monoterapi har ikke lenger noen plass i behandlingen av KLL.
- Fludarabin 40 mg /m2 p.o. eller 25 mg/m2 i.v. (i 100 mL NaCl 0,9 % over 30 minutter) eller 40 mg/m2 dag 1–3
- Syklofosfamid 250 mg/m2 p.o. eller 250 mg/m2 i 100 mL 5 % Glucose over 30 minutter dag 1–3 som gjentas hver 28. dag.
Dersom det er indikasjon for fludarabin-basert behandling bør den alltid gis som FCR-kurer; dvs. rituksimab eller annet anti-CD20 antistoff sammen med FC.
Dosen justeres etter følgende retningslinjer: Dersom pasienten på dag 1 av enhver kur (unntatt kur 1) har granulocytter <1,0 x109/L og/eller trombocytter <50 x 109/L (hematologisk toksisitet grad 3), utsettes kuren i opptil 2 uker og gis med 25 % dosereduksjon. Dersom lignende beinmargstoksistet oppstår til tross for 25 % dosereduksjon anbefales dosereduksjon til 50 % av utgangsdosen.
Fludarabin utskilles for en stor del gjennom nyrene. Dersom kreatinin-clearance er redusert til 30–60 mL/min anbefales en halvering av dosen. Hos pasienter med kreatinin-clearance <30 mL/min bør alternativ behandling vurderes.
Det anbefales PCP-profylakse og transfusjon med bestrålte blodprodukter, men ikke rutinemessig virus-profylakse, se kapittel Komplikasjoner, avsnitt "Infeksjonsprofylakse".
Det er teoretiske grunner til å gi fludarabin først; hemme reparasjon av syklofosfamidinduserte DNA-skader i tumorceller.
Dosejusteringer pga. redusert nyrefunksjon eller hematologisk toksisitet bør gjøres som anført i avsnittet om fludarabin.
Monoklonale anti-CD20 antistoffer
Rituksimab er et kimært mus-humant monoklonalt antistoff med spesifisitet for CD20. Det har vært brukt i kombinasjon med kjemoterapi med fludarabin (F), klorambucil, syklofosfamid (C), FC eller Bendamustin, og øker responsrate og progresjonsfri overlevelse i forhold til kjemoterapi alene i de undersøkelser der slik sammenlikning er mulig.
Ved kombinasjon med kjemoterapi gis rituksimab oftest en gang hver 4. uke sammen med kjemoterapien.
Infusjonsrelaterte bivirkninger er influenzaliknende symptomer, hypotensjon og bronchospasmer. Risiko for infusjonsrelaterte bivirkninger er størst ved første infusjon, ved stor tumormasse og høye lymfocyttall. Premedikasjon med iv kortikosteroid, paracetamol og antihistamin anbefales før første infusjon. Senere infusjoner går vanligvis greit. Se Felleskatalogen for forholdsregler.
Fludarabin, syklofosfamid, og rituksimab (FCR)
Ved første kur gir noen rituksimab dagen før cytostatika (dag 0) eller fordeler dosen på to dager. Ved senere kurer gis rituksimab på dag 1.
- Rituksimab gis i dose 375 mg/m2 ved første kur og 500 mg/m2 ved senere kurer.
- Fludarabin og syklofosfamid gis som anført ovenfor for FC. Gjentas hver 28. dag.
Det er vanlig å gi rituksimab før fludarabin og syklofosfamid til slutt.
Ved cytopenier anbefales justering av syklofosfamid og fludarabindosene som anført ovenfor. Det er ikke nødvendig å justere rituksimabdosen ved cytopenier.
Det anbefales PCP-profylakse og transfusjon med bestrålte blodprodukter men ikke rutinemessig virus-profylakse, se avsnitt Komplikasjoner, avsnitt "Infeksjonsprofylakse".
Obinutuzumab er et annen generasjons anti-CD20 antistoff godkjent i Europa for behandling av KLL i første linje kombinert med klorambucil. Beslutningsforum har gitt sin tilslutning til at obinutuzumab kan benyttes i Norge.
- Obinutuzumab gis i dose fast dose på 1000 mg. Ved første behandling er det anbefalt å gi en testdose på 100 mg og dessuten premedikasjon som skissert for rituksimab.
Ofatumumab er ikke lenger tilgjengelig.
Kortikosteroider
Ved immunmedierte cytopenier (anemi, trombocytopeni og/eller nøytropeni) er prednisolon peroralt førstevalg; vanligvis 1–2 mg/kg initialt. Nedtrapping skjer etter respons og må individualiseres. Se kapittel Autoimmune cytopenier.
Ved beinmargssvikt forsøker noen
- Høydose metylprednisolon (1 g/m2 iv i tre dager) gitt med 4 ukers intervall,
evt kombinert med - Rituksimab (375 mg/m2 iv dag 1, 8, 15 og 22)
Signalveishemmere
To kinasehemmere som hemmer signalering gjennom B-cellereseptoren er nylig markedsført i Norge.
Ibrutinib hemmer Brutons tyrosin kinase (BTKI). Det administreres p.o. i startdose 420 mg x 1 som kontinuerlig behandling og tåles vanligvis godt. Diare, tretthet og infeksjoner er de hyppigste bivirkningene. De fleste får økende lymfocytose den første tiden samtidig som glandelsvulst reduseres.
Acalabrutinib er annen generasjons BTKI. Det administrerers p.o. som kontinuerlig behandling; kapsler 100 mg 2 ganger dalig. Acalabrutinib forventes å få markedsførignstillatelse i Norge i 2021, og Metodevurdering forventes ferdigstillet i juni 2021.
Idelalisib er en PI3K hemmer som administreres p.o. 150 mg x 2 som kontinuerlig behandling med liknende effekt og bivirkningsprofil som ibrutinib, men inkluderer også leverpåvirkning og pneumonitt. Bivirkninger ses noe hyppigere ved idelalisib enn ibrutinib.
BCL-2 hemmer
Venetoklaks er en selektiv BCL-2-hemmer som administreres per oralt. Venetoklaks har effekt ved maligniteter med overekspresjon av BCL-2 slik som KLL.
Medikamentet tolereres realtivt godt. Den største utfordringen er utvikling av tumorlyse-syndrom ved oppstart og/eller doseøkning. Det anbefales derfor en gradvis opptrapping av dosen fra 20 mg daglig til 400 mg daglig over en 5 ukers periode og samtidig må man sørge for at pasientene har et høyt væskesinntak (2–2,5 L/daglig) og benytter urikosuriske medikamenter i hele opptrappingsfasen.
Komplikasjoner
Sist faglig oppdatert: 23.12.2021
Infeksjoner
Infeksjonsbehandling
Infeksjoner er den dominerende komplikasjonen ved KLL og den hyppigste KLL-relaterte dødsårsaken. Høy infeksjonsrisiko er relatert til nøytropeni som ledd i beinmargssvikt, behandling med kortikosteroider, hypogammaglobulinemi og defekt T-cellefunksjon.
Raskt innsatt empirisk antibiotikabehandling er avgjørende for prognosen ved bakterielle infeksjoner, og man bør starte intravenøs synergistisk bakterisid behandling (1. linjebehandling: betalaktam + aminoglycosid; 2. linjebehandling: karbapenem) hos enhver høyfebril/medtatt pasient uansett om patogenet er påvist eller ikke. Med økende bruk av purinanaloger, høydosert meylprednisolon, idelalisib og ibrutinib bør man også ta høyde for at opportunistiske infeksjoner kan foreligge (Pneumocystis jirovecii, HSV, CMV, EBV, Listeria monocytogenes og muggsopp). Virus-reaktivering er også rapportert ved behandling med ibrutinib (HBV), og det er også kommet rapporter om muggsopp-infeksjoner ved ibrutinib spesielt i kombinasjon med kortikosteroider (Robak et al., 2010; Wierda et al., 2010). Profylaktisk antibiotikabehandling ved residiverende bakterielle infeksjoner anbefales ikke i Norge, først og framst av hensyn til resistensutvikling. Effekten av slik behandling er ikke undersøkt i prospektive kliniske studier.
Immunglobulinbehandling
Hypogammaglobulinemi sees hos et stort flertall av pasientene (opptil 70 % i uselekterte materialer) og prevalensen øker med sykdomsvarigheten.
- Ved hyppige residiverende bakterielle infeksjoner (infeksjoner med kapselkledde bakterier) kan substitusjonsbehandling med immunglobulin være aktuelt hvis vaksinasjon (pneumokokkvaksine) ikke har ført fram og hypogammaglobulinemi foreligger (evidensgrad B).
Intavenøst humant immunglobulin (IVIG) 0,4 g/kg hver 3. uke er vist å ha effekt (Chapel et al., 1994; Griffiths et al., 1989). Dosen bør justeres på bakgrunn av Ig-kvantitering etter at substitusjonsbehandlingen er iverksatt. Lavere IVIG-dose 10 g/3. uke er rapportert å være effektivt (Jurlander et al., 1994). Subcutan administrasjon kan være et godt alternativ, og slik behandling kan administreres av pasienten sjøl etter opplæring.
På rett indikasjon reduserer IVIG hyppigheten av bakterielle luftveisinfeksjoner hos pasienter med KLL og hypogammaglobulinemi, men det diskuteres stadig om behandlingen kan anses kostnadseffektiv.
Vaksinasjon
Pasienter med KLL får suboptimal respons vurdert som stigning av antistofftiter etter vaksinasjon mot difteri, tyfus, parotitt, influenza, pneumokokker og haemophilus influenzae (Gribabis et al., 1994).
- Det er vanlig å anbefale pasienter med KLL influensa-vaksinasjon og vaksinasjon mot pneumokokker ved gjentatte pneumonier (evidensgrad D).
Nytten av en slik behandling hos pasienter med KLL er dårlig dokumentert (Sinisalo et al., 2003). Systematisk vaksinering av pasienter med KLL hvor infeksjoner ikke er eller har vært noe klinisk problem anbefales ikke. Det kan være hensiktsmessig å avklare den enkelte pasientens evne til å respondere på vaksinasjon ved å bestemme anti-pneumokokk-antistoff titer før og 8 uker etter vaksinasjon.
Infeksjonsprofylakse
- T-celledefekten som skyldes behandling med fludarabin gjør at profylaktisk behandling mot Pneumocystis jirovecii pneumoni (PCP) med trimetoprim-sulfametoxazol 1tbl daglig (eller 6–8 tabletter/uke fordelt på annen måte) anbefales under og minst seks måneder etter slik behandling, eventuelt til CD4+ celletallet i blod >0,4 x 109/L (evidensgrad D).
- PCP-profylakse anbefales også ved behandling med idelalisib.
Ved behandling med fludarabin og ibrutinib må en også være oppmerksom på muligheten for infeksjoner med virus (reaktivering) og for sopp-infeksjoner.
Profylakse mot soppinfeksjoner anbefales ikke rutinemessig pga. risikoen for resistensutvikling.
Autoimmune cytopenier
Sist faglig oppdatert: 23.12.2021
Autoimmune cytopenier forekommer langt hyppigere hos pasienter med KLL enn i en aldersjustert normalbefolkning. Autoimmun hemolytisk anemi (AIHA), immunmediert trombocytopeni (ITP) og erytroaplasi (Pure red cell aplasia, PRCA) forekommer med en insidens på henholdsvis 4–40 %, 1–2 % og <1 % (Hamblin, 2006). Omtrent dobbelt så mange pasienter har en positiv DAT under sykdomsforløpet enn de som utvikler AIHA. AIHA kan forekomme ved ubehandlet KLL, men det er velkjent at insidensen øker etter behandling både med fludarabin og klorambucil. Tillegg av syklofosfamid reduserer risiko for AIHA (Catovsky et al., 2007; Dearden et al., 2008). AIHA etter behandling kan oppstå uavhengig av DAT status før behandling, men ses hyppigere hos DAT+ pasienter.
- Autoimmune cytopenier behandles med Prednisolon 50 mg x 2 (eventuelt 1 mg/kg/d) i en uke, 25 mg x 3 i en uke og deretter avtrappende dosering alt etter effekt (evidensgrad C).
Når hemolysen er under kontroll trappes prednisolon langsomt ned over et par måneder. Ved manglende respons eller residiv av hemolysen under nedtrapping av prednisolon er det aktuelt å starte antileukemisk behandling.
Ved hemolyse som inntreffer under behandling seponeres behandlingen omgående, og det gis prednisolon som anført over. Dersom steroider ikke er tilstrekkelig for å kontrollere hemolysen kan kombinasjon med ciklosporin prøves, men i en slik situasjon vil det som regel være aktuelt å starte alternativ antileukemisk behandling.
Autoimmune cytopenier som opptrer etter behandling med fludarabin kan ofte være svært alvorlig og ende fatalt (Myint et al., 1995; Weiss et al., 1998). De aller fleste pasienter som utvikler AIHA etter fludarabin får på nytt hemolyse ved reeksposisjon for fludarabin. Rebehandling med fludarabin hos pasienter som har hatt AIHA etter fludarabinbehandling frarådes derfor.
- Rituksimab og rituksimab i kombinasjon med syklofosfamid er et aktuelt alternativ ved AIHA, erytroaplasi og andre immunmedierte komplikasjoner som ikke responderer på prednisolon (evidensgrad C).
Dokumentasjonen er basert på små studier studier (Ghazal, 2002; Gupta et al., 2002; Hegde et al., 2002).
Transformasjon til høymalignt lymfom
Sist faglig oppdatert: 23.12.2021
Richters transformasjon eller Richters syndrom er en klinisk patologisk betegnelse på rask utvikling av histologisk verifisert aggressivt lymfom hos en pasient med KLL. Det vanligste lymfomet ved Richters transformasjon er diffust storcellet B-celle lymfom (DLBCL), men andre varianter av lymfom er også beskrevet; spesielt Hodgkin lymfom (Österborg et al., 2009). Studier har vist at DLBCL ved Richters transformasjon kan være klonalt relatert til pasients opprinnelige KLL (ca. 80 %) eller ikke klonalt relatert til KLL (ca. 20 %); dvs. de novo DLBCL (Mao et al., 2007; Rossi et al., 2011). Overlevelse etter Richters transformasjon varierer mellom noen få uker til 15 år i ulike rapporter, men det synes å være enighet om at prognosen er dårligst i de tilfellene hvor DLBCL er klonalt relatert til KLL (Österborg et al., 2009). Når DLBCL ikke er klonalt relatert til KLL er prognosen som ved de novo DLBCL, og behandlingen bør være som ved de novo DLBCL.
Endret sykdomsbilde i form av rasktvoksende lymfeknutesvulst, kraftig økning av LD, B‑symptomer, abdominal symptomer og/eller ekstranodale manifestasjoner bør gi mistanke om transformasjon. Prolymfocytt-transformasjon karakteriseres av økende lymfocytose med karakteristisk morfologi og immunfenotype, splenomegali og beinmargssvikt. Diagnosen må verifiseres ved biopsi, immunfenotyping og eventuelt også cytogenetikk. PET-undersøkelse kan være til hjelp for å identifisere hvilke(n) lesjon(er) som er best egnet for biopsi.
Det anbefales at pasientene behandles etter de samme retningslinjene som gjelder for tilsvarende sykdommene uten assosiasjon til KLL.
- R-CHOP er et vanlig brukt regime ved DLCBL (evidensgrad C). EPOCH-(F)R er et alternativ (evidensgrad C). Dersom lymfomet ikke er klonalt relatert til KLL anbefales behandling som ved de novo DLBCL (evidensgrad D), mens det anbefales konsolidering med allogen stamcelletransplantasjon når lymfomet er klonalt relatert (evidensgrad D).
- R-CHOP kan også være et alternativ ved prolymfocytt leukemi i likhet med fludarabinbasert kjemoimmunoterapi (evidensgrad D).
Prognosen er dårlig ved transformasjon og rent palliativ behandling kan ofte være aktuelt.
Myelomatose
Sist faglig oppdatert: 21.12.2023
Anbefalinger:
- Diagnose og behandlingsindikasjon settes etter 2014-kriteriene, og krever biopsi av enten benmarg eller plasmacytom.
- Risikostratifisering av utredningen anbefales med FISH, ISS og R-ISS.
- Høydosebehandling med stamcellestøtte anbefales som en del av førstelinjebehandlingen hos pasienter under 70 år uten betydelig komorbiditet.
- VRd eller Dara-VTd induksjon anbefales
- For vurdering av tandem, konsolidering og vedlikehold, se underkapittel "Førstegangsbehandling av myelomatose".
- For pasienter over 70 år eller med betydelig komorbiditet, anbefales VRd.
- Det anbefales å endre behandling ved biokjemisk tilbakefall eller klinisk tilbakefall.
- Anbefalte behandlingslinjer: Velcade-Revlimid-dex => Daratumumab-Velcade dex => Karfilzomib-dex => Pomaolidomid-dex => Belantamabmafodotin
- Det anbefales zoledronsyre 4 mg i.v. hver 4. uke i 2 år. Hvis pasienten oppnår VGPR kan behandling avsluttes etter minst 1 års behandling. Deretter eventuelt re-oppstart ved skjelettprogresjon.
- Denosumab kan vurderes ved kontraindikasjon for zoledronsyre.
Diagnostiske kriterier
Sist faglig oppdatert: 21.12.2023
Diagnosen myelomatose bygger på biopsi fra benmarg eller biopsi fra en tumor. I tillegg vurderes om det foreligger tegn på organpåvirkning eller tilstedeværelse av myelomdefinerende biomarkører. Det skilles mellom plasmacytom, monoklonal gammopati av usikker betydning (signifikans) (MGUS), ulmende («smouldering») myelomatose og behandlingskrevende myelomatose. Diagnose og indikasjon for behandling bygger på kriterier publisert av International Myeloma Working Group i 2014 (Rajkumar et al., 2014) og er referert nedenfor.
Definisjon av behandlingskrevende myelomatose
≥10 % klonale plasmaceller i benmarg eller biopsiverifisert plasmacytom i bein eller ekstramedullært og minst ett myelomatosedefinerende kriterium.
Myelomatosedefinerende kriterier:
C Hypercalcemi. Albumin-korrigert Ca >0,25 mmol/l over referanseverdi eller > 2,75 mmol/l.
R Nyresvikt. Forhøyet kreatinin >177 µmol/l eller kreatinin clearance <40 ml/min (målt eller estimert). Biopsi anbefales ved tvil om relasjon mellom plasmacellesykdommen og nyresvikten.
A Anemi. Hb <10 g/dl eller 2 g/dl under laboratoriets referanseområde eller under pasientens individuelle normalverdi.
B Skjelettsykdom. En eller flere osteolytiske lesjoner påvist ved lavdose CT eller røntgen. Ved usikre funn bør undersøkelsen gjentas etter 3–6 måneder fremfor å starte behandling
Ved fravær av kriteriene A til B vil tilstedeværelse av ett av følgende tre kriterier også regnes som myelomatosedefinerende kriterium:
- ≥60 % klonale plasmaceller i beinmargen
- ratio serum kappa/lambda eller lambda/kappa ≥100
(høyeste lettkjedeverdi må være >100 mg/L) - ≥2 fokale lesjoner på MR
(undersøkelse kan begrenses til columna/bekken, men mister da 20 % av diagnosene)
Kommentarer
Bemerk at infeksjonstendens, polynevropati, osteoporose eller kompresjonsfraktur ikke er blant kriteriene da de betraktes som for uspesifikke. Husk også at spesielt anemi, hypercalcemi og nyresvikt må utredes med tanke på andre årsaker.
Størrelsen på CT og MR-lesjoner er satt til minst 5 mm. Hvis benmargsbiopsi viser <10 % klonale plasmaceller og det fortsatt er mistanke om myelomatose, må biopsien gjentas, eventuelt må det gjøres målrettet biopsi av en lesjon.
Definisjon av ulmende myelomatose
- Serum monoklonalt protein ≥30 g/l og/eller urin monoklonalt protein ≥500 mg per 24 t og/eller klonale plasmaceller i benmarg 10–59 %.
- Ingen myelomatosedefinerende kriterier og fyller ikke kriterier for AL-amyloidose.
Definisjon av MGUS
- <10 % klonale plasmaceller i benmarg.
- Tilstedeværelse av klonal markør i serum eller urin.
- monoklonal komponent i serum <30 g/l og urin monoklonalt protein <500 mg per 24t.
og/eller
-
- FLC-ratio <0,26 eller >1,65
- Fyller ikke noen myelomatosedefinerende kriterier
Solitært plasmacytom
Sist faglig oppdatert: 21.12.2023
(Omfatter også myelomer som vokser ekstramedullært ut fra ben.)
- Solitær lesjon i ben eller bløtvev forårsaket av klonale plasmaceller (biopsi).
- CT skjelett og MR columna og bekken viser ingen ytterligere lesjoner.
- Ingen myelomatosedefinerende kriterier oppfylt.
Pasientene inndeles i solitært plasmacytom, som ikke har klonale plasmaceller i benmarg og har en risiko for progresjon til myelomatose er 10 % i løpet av 3 år, og Solitært plasmacytom med minimal marginfiltrasjon, som har <10 % klonale plasmaceller i blod og har en risiko for utvikling av myelomatose på 60 % og 20 % i løpet av tre år for plasmacytom i hhv ben og bløtvev.
(Rajkumar et al., 2014; Soutar et al., 2004)
Plasmacelleleukemi
Sist faglig oppdatert: 21.12.2023
- Plasmaceller >5 % av leukocytter i blod (Fernández de Larrea et al., 2021).
Avgrensing mot andre relevante sykdommer
Sist faglig oppdatert: 21.12.2023
Mb Waldenström
Mb Waldenström er definert som påvisning av lymfoplasmacytisk lymfom i benmargen og sirkulerende monoklonalt IgM (se underkapittel om Waldenströms makroglobulinemi). Man ser imidlertid av og til IgM-myelomatose.
For å skille disse er følgende informasjon nyttig:
- MYD88 L265P-mutasjonen er bare til stede i Mb Waldenström
- Translokasjoner i kromosom 14 og osteolytiske lesjoner er bare til stede ved myelomatose. Ved IgM-myelomatose er t(11;14) den vanligste.
Hvis ikke noe av dette er til stede, må man basere seg på patologens vurdering hva sykdommen passer best med. Hvis konklusjonen er at det er myelomatose, men pasientens klonale plasmaceller er CD20-positive, er det teoretiske holdepunkter for at CD20-antistoffer kan være nyttig som en del av behandlingen.
AL Amyloidose
Se eget kapittel. Pasienter med MGUS eller myelomatose utredes og følges med følgende prøver for å fange opp potensiell samtidig AL amyloidose:
- NT-ProBNP, ALP, INR, U-albumin
Utredning av myelomatose
Sist faglig oppdatert: 21.12.2023
Klonale undersøkelser og immunglobuliner
Proteinelektroforese serum. En monoklonal komponent (M-komponent) fremstår som et skarpt ekstra bånd, vanligvis i gammasonen, men kan også sees i beta-sonen. Komponentens konsentrasjon i g/L bestemmes ved scanning basert på fargeintensitet i det ekstra båndet. Prosedyren for dette kan variere noe og må klareres med laboratoriet. Når det påvises m-komponent, må laboratoriet kvantitere denne. Det er ikke tilstrekkelig å beskrive kvalitativt (større/mindre/stabil), og det må gjøres typing av m-komponenten. Ved spørsmål om komplett respons, må det også gjøres immunfiksering. Noen sykehus bruker kapillærelektroforese som er mer sensitivt enn vanlig gel-elektroforese. Det må likevel understrekes at negativ kapillærelektroforese ikke er tilstrekkelig for komplett respons, det må uansett gjøres immunfiksering.
Konsentrasjon av IgG, IgA og IgM. Det er nyttig å vite om normale immunglobuliner er supprimert (immunoparese) fordi dette kan medvirke til infeksjonstendens.
Serum frie lette kappa- og lambda-kjeder (S-FLC). Økning av kappa eller lambdakjeder i serum sees hos ca. 80 % av myelomatosepasienter som produserer komplett immunglobulin og hos 100 % av de med lettkjedesykdom. Bestemmelse av lette kjeder i serum brukes ofte som alternativ til urinelektroforese, men det gir ikke alltid tilsvarende svar.
Økning av begge lettkjedetyper skyldes ikke myelomatose, men kan ses ved nyresvikt med redusert utskillelse eller en polyklonal immunrespons. FLC-ratioen endres noe ved nyresvikt. Følgende referanseverdier gjelder for pasienter med nyresvikt (Long et al., 2022):
- eGFR 45-59 ml/min/1.73m2: 0.46 – 2.62 mg/L
- eGFR 30-44 ml/min/1.73m2: 0.48 – 3.38 mg/l
- eGFR <30 ml/min/1.73m2: 0.54 – 3.30 mg/l
Alle kriterier som baserer seg på lette kjeder (responser, definisjon av MGUS/ulmende myelomatose/behandlingstrengendemyelomatose, progresjon) er basert på Binding Site-metoden. Andre metoder gir forskjellige svar. Vi anbefaler da at denne metoden brukes, inntil man har funnet en felles standard.
Immunfiksasjon/elektroforese urin. Immunfiksering for påvisning, typing og kvantitering brukes på samme måte i urin som i serum. Sensitivitet for immunfiksering er ca. 10 mg/L. Urinelektroforese er relativt arbeidskrevende og kan i rutinepraksis utelates hvis pasienten har serum M-komponent eller frie lette kjeder i serum som kan følges. Men urinelektroforese med døgnsamling er fortsatt den standardundersøkelse i responskriteriene og derfor i alle myelomatosestudier. Spot- eller morgenurin har bare betydning ved påvisning av komplett respons, og kan ikke brukes til annen responsvurdering.
Bildediagnostikk ved myelomatose
Sist faglig oppdatert: 21.12.2023
Lavdose CT er standardutredning av skjelett ved myelomatose (Wolf et al., 2014). Lavdose CT gir en stråledose på ca. 4,1 mSv, og krever ingen kontrast. Man bør gjøre lavdose CT ved oppstart av hver behandling, hvis det ikke er tatt de siste 6 månedene.
MR fremstiller skjelettets bløtvev på en bedre måte enn CT, og vil gi bedre informasjon om intraspinale forhold og spørsmål om truende tverrsnittskade. MR har høy sensitivitet for myelomer, men kan ikke brukes til vurdering av beindestruksjon og frakturfare. MR gir ingen strålebelastning. Kartlegging før strålebehandling bør gjøres ved MR da det ofte er snakk om bløtdelsforandringer. MR-lesjoner er et av kriteriene som kan definere behandlingskrevende myelomatose. MR skal gjøres ved utredning og oppfølgning av solitært plasmacytom utgående fra skjelettet, og for å sette diagnosen ulmende myelomatose. MR trengs ikke hvis pasienten har behandlingstrengende myelomatose ved andre kriterier.
Skjelettscintigrafi brukes ikke.
FDG-PET-CT brukes i følgende situasjoner:
- Obligat ved oppfølging av non-sekretorisk myelomatose
- Obligat ved utredning og oppfølging av ekstramedullært solitært plasmacytom
- Kan brukes som prognostisk undersøkelse etter HMAS, før eventuelt vedlikehold, og oppfølging årlig av pasienter med positiv PET fra denne undersøkelsen.
Makrofokal sykdom
Sist faglig oppdatert: 21.12.2023
Makrofokal myelomatose er en subgruppe med utbredt skjelettsykdom men med lav mengde plasmaceller i benmargen. Gruppen har ikke anemi, nyresvikt eller hypercalcemi. Pasientene har ofte plasmaceller under 10% i benmargen, og det trengs da en målrettet biopsi for å stille diagnosen.
Cytogenetiske forandringer ved myelomatose er svært hyppige. Forandringer som innbefatter tungkjedeimmunglobulingenet på kromosom 14 regnes som primære og har i de fleste tilfeller patogenetisk betydning. MGUS og myelomatose har de samme cytogenetiske forandringene, men andelen pasienter som har slike forandringer er større ved myelomatose. Cytogenetikk kan ikke brukes diagnostisk, men har prognostisk og dels terapeutisk betydning.
Hypodiploiditet, t(4;14), t(14;16), t(14;20), gain1q, del 1p, del 13 og/eller del 17 ved diagnosetidspunkt indikerer dårlig prognose, mens hyperdiploiditet gir god prognose. Den beste prognostiske indeksen får man ved å kombinere FISH med LD og ISS, og dette er gjort i R‑ISS (revidert ISS-score). Forekomsten av cytogenetiske avvik fordeler seg omtrent slik hos pasienter i Norge: t(4;14): 15 %, del 13: 50 %, del 17: 15 %, t(11;14): 15 %.
Del17p og gain1q tilkommer i forløpet av myelomatose. Det anbefales derfor å gjenta FISH ved tilbakefall hos pasienter som ikke tidligere har påvist disse.
Metode for cytogenetisk undersøkelse
Fluoriserende in situ hybridisering (FISH) er standardmetode. Det er ikke nødvendig å gjøre karyotyping, men man mister da muligheten til å påvise hyperploiditet. De tre avvikene som inngår i R-ISS [t(4;14), t(14;16) og del 17p utgjør sammen med t(11;14) et minimum. Helst bør også kromosom-1-endringer og t(14;20) gjøres.
Biobank og FISH
Det anbefales at det sendes prøver til biobank. Det kan gjøres enten ved å sende tillab for prøvetaking og spesiell hematologi ved St. Olavs hospital, eller ved å kontakte Oslo myelomatosesenter. Ved å sende prøver dit får man svar tilbake på FISH for del17, t(4;14), t(14;16), t(11;14), t(14;20) og gain1q som er de viktigste endringene av prognostisk betydning. Rekvisisjonsskjema og samtykkeskjema sendes samlet med prøvene. Disse skjemaene finnes hos St. Olavs hospital: rekvisisjoner og samtykke.
Anbefalinger for utredning
Sist faglig oppdatert: 21.12.2023
Standardutredning blod og urin
- Serumelektroforese med kvantitering av M-komponent. Frie lette kjeder.
- Hb, hvite med diff. telling, trombocytter, kreatinin, estimert GFR, Calcium eller ionisert Calcium, albumin, β2mikroglobulin, LD, IgG, IgA, IgM.
- NT-Pro-BNP, Troponin T, ALP, og Urin-albumin. Blodutstryk. PTH-måling bør vurderes ved hypercalcemi for å utelukke primær hyperparathyreoidisme.
Nivåer og rekkefølge på utredning
Sist faglig oppdatert: 21.12.2023
Lavrisikoutredning (MGUS mer sannsynlig diagnose)
Hvilke pasienter:
- Normal lettkjederatio, IgG m-komponent <15 g/l, ingen symptomer.
Hva skal gjøres:
- Blod- og urinprøver (se avsnitt MGUS)
- Anamnese på symptomer fra myelomatose og/eller amyloidose
- Ikke benmarg eller radiologisk undersøkelse
Høyrisikoutredning
Hvilke pasienter.
- Abnormal lettkjederatio eller
- IgG m-komponent >15 g/l eller
- IgA m-komponent eller
- Plasmacytom eller
- Mistenkt relaterte symptomer
Hva skal gjøres:
- Blod- og urinprøver (se Oppfølging av MGUS, ulmende myelomatose og plasmacytomer)
- CT myelomatose
- Benmargsutstryk og benmargsbiopsi
MR helkropp gjøres (ev. columna og bekken hvis ikke tilgjengelig) hos pasienter med >10 % plasmaceller i biopsi, men som ikke fyller kriterier for behandlingskrevende myelomatose.
Fullutredning
Hvilke pasienter:
- Pasienter som har påvist behandlingskrevende myelomatose
- Nyhenviste pasienter med «sikker» behandlingskrevende myelomatose
- Pasienter med påviste plasmacytomer med klonal sykdom i benmargen
- Pasienter med ulmende myelomatose
Hva skal gjøres:
- Utredning som under «høyrisikoutredning»
- Virusserologi for HBV, HCV og HIV
- FISH av benmarg
- Biobanking om mulig
Andre undersøkelser:
Flowcytometri: Fordi minimal restsykdom (MRD)-vurdering ved flowcytometri har blitt mer vanlig, er det ønsket fra flowcytometrimiljøet at det tas en baseline flowcytometri av pasienter i førstelinje. Hvorvidt dette kan gjøres må avklares lokalt.
PET ved diagnose, og hvordan dette endrer seg etter behandling, gir prognostisk informasjon, og kan være aktuelt på enkelte sykehus.
Oppfølging av MGUS, ulmende myelomatose og plasmacytomer
Sist faglig oppdatert: 21.12.2023
MGUS
Det anbefales oppfølging hos fastlege etter 6 måneder og deretter årlig. Følgende prøver bør følges: Hb, kreatinin, calcium, NT-pro-BNP, ALP, S-elektroforese, frie lette kjeder, U-albumin.
Lav terskel for radiologiske undersøkelser, og tilbakehenvisning ved dynamikk i prøvene. Husk symptomer og funn tydende på AL-amyloidose, viktigst er albuminuri og forhøyet NT-pro-BNP.
Plasmacytomer etter behandling og ulmende myelomatose
Det anbefales oppfølging ved hematologisk poliklinikk hver 3. måned med standard blod- og urinprøver.
I tillegg, radiologisk oppfølging:
- Solitært ekstramedullært plasmacytom: FDG-PET/CT årlig i 5 år
- Solitært plasmacytom utgående fra skjelett: MR helkropp årlig i 5 år
- Ulmende myelomatose med én MR lesjon: Alternerende MR helkropp og CT skjelett hver 6. mnd
- Ulmende myelomatose uten MR-lesjoner: Årlig MR helkropp. Kan avsluttes etter 5 år ved stabile forhold.
Stadieinndeling og prognose
Sist faglig oppdatert: 21.12.2023
Det fins mange prognostiske markører. ISS er mest brukt og kan anvendes hos alle pasienter, også ved høydosebehandling og nedsatt nyrefunksjon. R-ISS (revised-ISS) er utarbeidet for pasienter behandlet initialt med IMIDs og proteasomhemmere.
ISS og FISH skal tas hos alle pasienter, og er et kvalitetsmål i kreftregisterets årlige rapport for B-cellesykdommer.
Stadium I | S-β2 mikroglobulin <3,5 mg/l og S-albumin ≥35 g/l |
Stadium II | Verken I eller II |
Stadium III | S-β2 mikroglobulin ≥5,5 |
R-ISS stadium I | ISS stadium I og normal LD og ikke høyrisiko FISH (del17p, t(4;14), t(14;16) | 5 års overlevelse 82 % |
R-ISS stadium II | Verken R-ISS I eller R-ISS III | 62 % |
R-ISS stadium III | ISS III og samtidig høy LD eller pos del17p, t(4;14) eller t(14;16). | 40 % |
Behandling
Sist faglig oppdatert: 21.12.2023
Nye Metoder
Nye metoder er et nasjonalt system for innføring og utfasing av metoder i spesialisthelsetjenesten. Det er de regionale helseforetakene som eier Nye metoder. Stortinget har vedtatt å lovfeste prioriteringskriteriene og at de regionale helseforetakene skal sørge for et felles system for å beslutte hvilke metoder som kan tilbys i spesialisthelsetjenesten. Handlingsprogrammene omtales medikamenter og kombinasjoner på bakgrunn av evidens fra kliniske studier, og det informere som finansiering. Et handlingsprogram kan ikke anbefale legemidler eller andre metoder som ikke har offentlig finansiering.
Metoder som er til vurdering skal som hovedregel ikke tas i bruk i norske sykehus. De regionale fagdirektørene har utarbeidet to prosedyrer for unntak fra bestemmelsen, en for grupper av pasienter og en for enkeltpasienter. Se Unntaksordning (nyemetoder.no).
Medikament | Indikasjon | Administrajon | Sak i NyeMetoder |
|---|---|---|---|
d | 1. linje ikke HMAS | Sc + po | Refundert |
Kar/d
Kar/len/d Kar/dara/d | Fra 1. relaps
Fra 1. relaps Fra 1. relaps | iv 30 min | Refundert Karfilzomib (Kyprolis) og deksametason (nyemetoder.no) Karfilzomib (Kyprolis) (nyemetoder.no) Daratumumab (Darzalex), karfilzomib (Kyprolis) og deksametason (nyemetoder.no) |
Elo/len/d Elo/pom/d | Fra 1. relaps Fra 2. relaps | iv 30 min | Refundert fra 3.relaps Under vurdering |
Ixa/len/d | Fra 1. relaps | Po | Refundert fra 3. relaps |
Dara Dara/V/d Dara/VTd | 2. relaps 1. relaps 1. linje ikke HMAS 1. relaps 1. linje ikke HMAS 1. linje før HMAS | sc | Refundert Kan brukes Under vurdering Refundert Under vurdering Under vurdering |
Len | Vedlikehold etter høydosebehandling | Po | Refundert |
Len/d | 1. linje ikke HMAS | Po | Refundert |
Pom/d Pom/Vel/d | 2. relaps 1. relaps | Po | Refundert Kan brukes |
Isa/Pom/d Isa/Kar/d | 2. relaps 1. relaps | i.v. 4-5-timer | Under vurdering Refundert |
Teclistamab | 3.relaps | s.c. | Under vurdering |
Cilta-cel | 3.relaps | i.v. | Under vurdering |
Belantamabmafodotin | 5. linje | i.v. 30min | Refundert |
Xgeva (Denusomab) | Forebyggende mot skjeletthendelser | Sc | Refundert |
Status per 17.10.2023. Oppdateringer kan sjekkes på nyemetoder.no
| Forkortelse | Forklaring |
|---|---|
D eller Dex | Dexametason |
Dara | Daratumumab |
Elo | Elotuzumab |
Isa | Isatuximab |
Ixa | Ixazomib |
Kar | Carfilzomib |
Len | Lenalidomid (Revlimid) |
Len-Dex | Lenalidomid-Deksametason |
M | Melfalan |
MPV | Melfalan-Prednisolon-Bortezomib |
P | Prednisolon |
Pan | Panobinostat |
Pom | Pomalidomid |
R | Revlimid |
V | Bortezomib (Velcade) |
VMP | bortezomib-melfalan-prednison |
VRD | bortezomib-lenalidomid-deksametason |
VTD | bortezomib-thalidomid-deksametason |
Generelt om behandlingsdoser, dosereduksjoner og seponering
Sist faglig oppdatert: 21.12.2023
Behandling av myelomatose har i de aller fleste tilfeller en kontinuerlig intensjon, og pasienter vil stå på behandling store deler av sitt gjenværende liv. På den ene siden fordrer kontinuerlig behandling god toleranse; pasienter skal tåle behandlingen de står på. På den andre siden er det viktig at pasienter som tåler behandlingen godt, får den behandlingen de har nytte av. Toleranse hos den enkelte pasienten kan være vanskelig å forutse. For å ivareta både pasientens mulighet for maksimal helsegevinst, og pasientens behov for et godt liv, foreslår vi derfor denne systematiske tilnærmingen.
Startdosene i et regime gis som det ble gitt i studien som ligger til grunn for dokumentasjonen. Dette er regimene som er oppført her i handlingsprogrammet, i Felleskatalogen, og som ligger til grunn for markedsføringstillatelsen og avgjørelser i Beslutningsforum.
Vi oppfordrer til lav terskel for dosereduksjon. Pasientene skal tolerere behandlingen, og gjør de ikke det skal dosen reduseres. Ved tvil er det bedre å gå ned i dose enn å la være å behandle. Dette innebærer selvfølgelig bruk av skjønn. Det er viktig at man tilstreber å prøve lavere doser fremfor å stoppe behandlingen, hvis ikke dette er strengt nødvendig.
Når man er på laveste dose og medikamentet fortsatt ikke tolereres, må dette seponeres. Hvis det finnes alternativer, bør ikke medikamentet restartes.
Førstegangsbehandling av myelomatose
Pasienter <70 år
- Pasienter <70 år tilbys høydose kjemoterapi med autolog stamcellestøtte (HMAS) med mindre det foreligger kontraindikasjoner for dette. Utvalgte pasienter med høy motivasjon >70 år kan også vurderes for dette.
Behandlingsrelatert mortalitet ved HMAS er lav selv om bivirkninger kan være betydelige.
Behandlingen omfatter 5 faser: 1) Induksjonsbehandling, 2) Autolog stamcellehøsting 3) HMAS 4) Konsolidering og 5) Vedlikeholdsbehandling.
Induksjonsbehandling og høsting. Det er naturlig å bruke induksjonsbehandling som gir den beste responsen, det vil si kombinasjoner av proteasomhemmere og imider. I retningslinjene anbefales 4 sykluser VRd eller Dara-VTd. Dara-VTd har gode resultater i en randomisert studie mot VTd. VTd er mer toksisk enn VRd, spesielt mtp nevropati. Griffin-studien har vist bedre effekt av Dara-VRd enn av VRd. Dara-VRd er i bruk i mange land, inkludert Sverige, og anses som det beste regimet. VRd anbefales nå med Velcade gitt ukentlig dag 1 og 8.
- Fordeler med VRd: Billigere enn Dara-VTd. Bedre tolerert.
- Fordeler med Dara-VTd: Sannsynligvis bedre effekt. EMA-godkjent.
- Fordeler med Dara-VRd: Den beste behandlingen
Autolog stamcellehøsting bør foregå kort etter induksjonsbehandlingen; helst tilsvarende tidspunkt på neste syklus. Stamceller høstes fra perifert blod etter behandling med Cyklofosfamid iv og G-CSF etter lokale retningslinjer. Høstingen kan gjøres uten cyklofosfamid pga potensielt alvorlige bivirkninger med cyklofosfosfamid, men cyklofosfamid gir noe høyere suksessrate.
Det høstes minimum 2 x 2,0 x 106CD34+ celler/kg kroppsvekt per HMAS, og for 3 HMAS hvis mulig.
HMAS. Etter induksjonsbehandlingen og høstingen gis Melfalan iv. Det bør være kortest mulig tid mellom høsting og melfalan. Reinfunderte stamceller skal være minimum 2,0 x 106 CD34+ celler/kg kroppsvekt. Dobbel (tandem) HMAS har vist forbedret overlevelse i hele populasjonen, og kan vurderes hos alle som tålte første HMAS godt. Tandem HMAS anbefales til alle pasienter med cytogenetisk høyrisk, pasienter med progresjon under induksjon og pasienter med plasmacelleleukemi. Hvis ingen reduksjon av klonale markører på første HMAS skal det ikke gis ny HMAS (unntatt hvis pasienten allerede er i CR etter induksjon)
Konsolidering: Konsolideringsbehandling er å fortsette med kurer tilsvarende induksjonskurene etter HMAS. Studiedata har vist forbedring av progresjonsfri overlevelse, og er ansett som standard behandling. Vi anbefaler derfor 2 nye sykluser med samme regime som konsolidering etter HMAS. Velcade kan og bør utelates hvis pasienten har signifikant nevropati.
Vedlikeholdsbehandling etter HMAS. En metaanalyse på 1208 pasienter med vedlikeholdsbehandling med lenalidomid etter HMAS (McCarthy et al., 2017) basert på 3 randomiserte studier, 2 med placebokontroll og en uten viste median totaloverlevelse >2 år lengre i behandlingsarmen. En engelsk større studie har senere vist tilsvarende resultater (Jackson et al., 2019). Det er økning av sekundære maligniteter i lenalidomidarmen, men dette slår ikke ut på dødeligheten. Det er farligere å ha myelomatose. Intensjonen i studien var behandling inntil progresjon. Pasienter sluttet imidlertid av ulike grunner og median tid på lenalidomid var 28 måneder.
På bakgrunn av disse studiene anbefales oppstart av vedlikeholdsbehandling med lenalidomid 3 måneder etter HMAS.
Anbefalt induksjonsbehandling
VRd
Bortezomib 1,3 mg/ m2 sc dag 1,8.
Lenalidomid 25 mg dag 1–14 fulgt av 7 dagers pause.
Dexamethason 40 mg po dag 1,4,8,11.
Gjentas hver 21. dag, i alt 4 ganger.
eller
Dara-VTd (28-dagerssykluser)
Daratumumab 1800 mg sc ukentlig i syklus og 1 og 2, annenhver uke i syklus 3, 4 og konsolidering
Bortezomib 1,3 mg/ m2 sc dag 1,4,8,11.
Thalidomid 100 mg daglig
Dexamethason 40 mg po dag 1,8,15,22.
Gjentas hver 28. dag, i alt 4 ganger.
eller
Dara-VRd (21-dagerssykluser)
Daratumumab 1800 mg sc ukentlig i syklus og 1 og 2, annenhver uke i syklus 3, 4 og konsolidering
Bortezomib 1,3 mg/ m2 sc dag 1,8.
Lenalidomid 25mg dag 1-14 fulgt av 7 dagers pause
Dexamethason 40 mg po dag 1,8,15
Gjentas hver 21. dag, i alt 4 ganger.
Ved eGFR<30 doserer man lenalidomid 15 mg daglig. Andre medikamenter doseres på samme måte.
VCd (aktuelt hvis imider er kontraindisert)
Bortezomib 1,3 mg/m2 sc dag 1,4,8,11.
Cyklofosfamid 900 mg/m2 dag 1
Dexamethason 40 mg po dag 1,4,8,11.
Gjentas hver 21. dag, i alt 4 ganger
CRd (aktuelt hvis bortezomib er kontraindisert -28dagerssykluser
Cyklofosfamid 500 mg dag 1 og 8
Revlimid 25 mg dag 1–21
Dexamethason 40 mg po dag 1,8,15,22
Perifer stamcellehøsting
Cyklofosfamid 2 g/m2 iv. (Kan utelates, se tekst ovenfor)
Uromitexan 800 mg/m2 x 4 (uroproteksjon) samme dag som cyklofosfamid.
G-CSF etter lokale retningslinjer.
Plerixafor kan brukes til pasienter med sterkt reduserte benmargsreserver.
Stamcellehøsting når CD34+ celler >20 x 106/l.
Høydosebehandling med autolog stamcellestøtte
- Melfalan 200 mg/m2. Ved eGFR <30 ml/min reduseres Melfalandosen til 140 mg/m2. Stamcellestøtte med minimum 2,0 x 106 CD34+ celler/kg.
Resultater fra både en randomisert studie (Cavo et al., 2020) og fra en metaanalyse (Cavo et al.) viser en overlevelsesgevinst for hele populasjonen ved tandemtransplantasjon vs enkel transplantasjon. Man kan derfor vurdere dette hos alle pasienter som hadde god toleranse for den første transplantasjonen. For pasienter med cytogenetisk høyrisiko, progresjon under induksjon eller plasmacelleleukemi er tandem anbefalt, hvis man responderer på den første. En eventuell transplantasjon nr. 2 gjøres ca. 3 måneder etter den første, med den samme dosen melfalan.
Konsolideringsbehandling
Vi anbefaler 2 konsolideringssykluser med det samme regimet etter HMAS. Konsolidering startes senest 3 måneder etter HMAS, gjerne tidligere hvis pasienter forventes å progrediere raskt.
Vedlikeholdsbehandling
- Lenalidomid vedlikeholdsbehandling etter HMAS har vist god effekt og anbefales. Behandlingen gis 21 av 28 dager, med 10mg daglig de første 3 syklusene og økning til 15mg hvis god toleranse. Man kan bruke pegfilgrastim hvis nøytropeni er et problem. Startes rett etter konsolidering, eventuelt 3 mnd etter HMAS. Hvis pasientene kommer fra konsolidering og tåler 25mg lenalidomid, kan man gå rett på 15mg.
Pasienter > ca. 70 år
DRd er den beste behandlingen i denne situasjonen, foran VRd og DVMP. DRd og D-VMP er under vurdering av beslutningsforum. VRd har vært i bruk i flere og må anses som førstevalget inntil disse vurderingene foreligger. Vi anbefaler at VRd gis som beskrevet under, dvs i 28-dagerssykluser, ikke 21. Hvis DRd godkjennes av beslutningsforum, vil dette være den beste behandling.
VRd (Durie et al., 2017)
Bortezomib 1,3 mg/m2 sc dag 1,8,15.
Lenalidomid 25 mg dag 1–21 fulgt av 7 dagers pause.
Dexamethason 40 mg po dag 1,8,15,22 (20mg hvis >75 år)
Gjentas hver 28. dag, 8 ganger, deretter kontinuerlige sykluser med Rd.
D-Rd
Daratumumab 1800 mg sc ukentlig 2 første sykluser.
Daratumumab hver 2. uke i syklus 3–6. Deretter 1 gang per syklus
Revlimid 25 mg dag 1–21
Dexametason 40 mg po dag 1,8,15,22 (20mg hvis >75 år)
Gjentas hver 4. uke til progresjon
D-VMP (Mateos et al., 2018)
Daratumumab 1800 mg sc ukentlig første syklus.
Daratumumab hver 3. uke 2.–9. syklus. Deretter 1 gang per syklus.
Bortezomib dag 1,4,8,11 og dag 22,25,29,32 i første syklus.
Bortezomib dag 1,8,22,29 i 2.–9. syklus.
Melfalan 6 mg/m2 og Prednisolon 60 mg/m2 dag 1–4 i 9 sykluser.
Gjentas hver 6. uke. Fra syklus 9 kun daratumumab.
For skrøpelige pasienter kan man starte med lenalidomid 15 mg, og etter 8 sykluser bør dexametason avsluttes og lenalidomid reduseres til 10 mg.
PFS øker ved kontinuerlig behandling, og er et alternativ hvis toleransen er god.
Alternativer som kan brukes hvis primæranbefalingen ikke kan følges.
Plasmacelleleukemi
Primær plasmacelleleukemi er en sjelden og aggressiv variant av plasmacellesykdommen. Den er definert ved plasmacelletall >5 % i blod hos en pasient som oppfyller kriteriene for myelomatose, og som ikke har fått behandling tidligere. Tellingen må gjøres i blodutstryk. Behandling i førstelinje er induksjonsbehandling med KRd, tandem HMAS og dobbelt vedlikehold med bortezomib og lenalidomid. Allogen stamcelletransplantasjon kan være aktuelt for utvalgte pasienter.
Allogen stamcelletransplantasjon
Allogen stamcelletransplantasjon med full eller redusert (RIC) kondisjonering har fortsatt høy mortalitet, men har et potensiale for kurasjon. Andel som kureres er lav, det er vanskelig å få frem sikre tall. Allogen stamcelletransplantasjon har vært gitt som primærbehandling eller i sekvens etter HMAS eller som tilbakefallsbehandling. Studiene viser varierende resultater som er oppsummert i en konsensusartikkel (Giralt et al., 2015). Det er ikke holdepunkter for at dårlig-prognose cytogenetikk eller klinisk aggressiv myelomatose gjør det bedre med allogen stamcelletransplantasjon. Da allogen stamcelletransplantasjon likevel har et potensiale for god effekt er det aktuelt å bruke dette hos pasienter med svært dårlig prognose. Den nasjonale transplantasjonsgruppen støtter denne nye anbefalingen.
Anbefaling:
Allogen stamcelletransplantasjon kan gis til pasienter som fyller følgende kriterier:
- Alder <55 år
- Aggressiv sykdom definert som progresjon før HMAS eller innen 6 måneder etter HMAS, eller plasmacelleleukemi
- Minst VGPR på andrelinjebehandling når man går til allogen stamcelletransplantasjon
Behandling ved nyresvikt
Thalidomid, bortezomib, bendamustin, carfilzomib, pomalidomid og daratumumab kan brukes uten dosereduksjon (begrenset erfaring ved eGFR <10 ml/min/1,73 m2). Revlimid gis i en dose på 15 mg daglig hvis eGFR <30, og i 25 mg hvis >30, men krever initialt tett oppfølging av kreatinin og celletall.
Pasienter med nyresvikt har dårligere prognose enn pasienter uten nyresvikt. Det er derfor viktig å behandle pasienter med nyresvikt minst like godt som pasienter uten nyresvikt, og unngå unødvendig dosereduksjon. Man bør altså tilstrebe triplettbehandlinger også på disse. Hvis man har startet med dosereduksjon bør denne forsøkes titrert opp ved god toleranse.
Påvirket nyrefunksjon sees hos ca. 50 % av pasientene på diagnosetidspunktet eller i sykdomsforløpet. Den vanligste årsaken er toksisk effekt av monoklonale lette kjeder i nyrenes proximale tubuli («myelomnyre»). Andre årsaker er dehydrering, infeksjoner, hyperkalsemi, nefrotoksiske medikamenter, hyperurikemi, hyperviskositet og amyloidose. Hvis behov for dialyse er både hemodialyse og peritoneal dialyse aktuelle alternativer. High-Cutoff-filter har vist bedre nyreoverlevelse etter 12 måneder, og anbefales derfor ved dialysebehov ved akutt nyresvikt der korreksjon av hyperkalsemi og hypovolemi ikke har gitt rask bedring av nyrefunksjonen.
Nyretransplantasjon kan gjøres ved langtkommen nyresvikt hvis nyrene er den dominerende/eneste sykdomsmanifestasjon og leveutsiktene ellers er gode.
Anbefaling:
- Hydrering som gir diurese >3 liter/døgn.
- Behandling av utløsende faktorer, spesielt hyperkalsemi og hyperurikemi.
- Nefrotoksiske medikamenter (aminoglykosider, NSAID) seponeres.
- Medikamenter: Alle myelomatosemedikamenter kan brukes
- Hvis pasienten er i dialyse bør man bruke High Cutoff-filter.
Behandling av tilbakefall
Sist faglig oppdatert: 21.12.2023
Når skal man starte behandling ved tilbakefall
Progressiv sykdom defineres som 25 % økning av M-komponenten i serum eller urin, i differansen mellom lette kjeder (dFLC) eller økning i plasmacelleandelen i benmargen (se tabell 8.2). Det finnes to typer indikasjoner for behandling av tilbakefall.
Klinisk tilbakefall: nye eller økte osteolytiske eller bløtvevslesjoner, forverring av hypercalcemi, anemi eller nyresvikt. Hyperviskositet.
Progressiv sykdom (se over og i tabell 8.2): Kort fortalt økning på 25 % i en eller flere klonale parametere, med et minimum på 5 g/l (s-m-komponent), 200 mg/døgn (u‑m‑komponent), 100 mg/l (dFLC), eller 10 % (benmarg) (Kumar et al., 2016).
Klinisk tilbakefall skal alltid behandles. I de fleste tilfeller bør også biokjemisk tilbakefall behandles. Det er publisert data som viser at det går bedre med pasienter som behandles på biokjemisk tilbakefall (Goldman-Mazur et al., 2023).
Aspekter og prinsipper for valg av behandling
- Alder
- Performance status
- Komorbiditet (organfunksjon, benmargsfunksjon, polynevropati mm.)
- Preferanse iv/sc versus tabletter
- Refraktæritet for behandling
- Toksisitet
Viktige prinsipper for tilbakefallsbehandling er følgende:
- Behandle paraproteinrelaps / progressiv sykdom
- Bytt til eller legg til ny virkningsmekanisme.
- All behandling er i utgangspunktet kontinuerlig, selv om man dosereduserer og deretter seponerer hvis manglende toleranse.
2. gangs HMAS
Dokumentasjonen for 2. gangs HMAS er relativt svak. Det er vist gevinst av HMAS vs peroral cyklofosfamid (Cook et al., 2014), og det er også teoretisk og erfaringsbasert grunnlag for å anbefale ny HMAS ved tilbakefall. Deler av tilnærmingen er likevel annerledes. HMAS er også aktuell ved tilbakefall hvis dette ikke er gjort før. 2. gangs HMAS har blitt mindre og mindre vanlig både i Europa og USA.
- Man bør vurdere en annen induksjonsbehandling. Bakgrunnen for dette valget er det samme som for hvordan man velger annen tilbakefallsbehandling, og dette forklares nedenfor.
- Da dokumentasjonen for ny HMAS er svak, bør ikke dette valget frata pasienter muligheten til godt dokumentert tilbakefallsbehandling. Derfor bør behandlingen gitt som induksjonsbehandling kontinueres etter HMASen. Pasientene bør altså få samme behandling på kontinuerlig basis som tilbakefallspasienter uten HMAS, men med et tillegg av HMAS etter 4 sykluser.
- Hvilken behandling man bruker som induksjon og kontinuerlig behandling, velges på samme grunnlag som annen tilbakefallsbehandling.
Råd for valg av regimer
(for dosering, se "Mer detaljer om behandlingsregimer" lenger ned i dette kapittelet)
Valg av regimer vil avhenge av flere faktorer, der de viktigste er overlevelsesgevinst i studier og hva pasienter tidligere har fått. Basert på konsensus, anbefales for en «standardpasient» følgende:
Førstelinje: Basert på bortezomib og lenalidomid
Andrelinje: Basert på daratumumab
Tredjelinje: Basert på carfilzomib
Fjerdelinje: Basert på pomalidomid
Femtelinje: Belantamab mafodotin
Anbefalt rekkefølgen på behandlingslinjer VRd => DVd => KRd => Pd => Belantamab.
Man kan vurdere å kombinere Kd eller Pd med bendamustin eller cyklofosfamid, men dokumentasjonen er dårlig
Det er også økende oppmerksomhet rundt opportunistiske infeksjoner ved bruk av bendamustin, og disse pasienter bør få Bactrim-profylakse.
En optimal rekkefølge hvis alt EMA-godkjent hadde finansiering ville vært DRd /DVRd-Len => KdD/Isa-Kd => EloPd => BCMA-rettet behandling
Venetoclax gir høye responser og god PFS i pasienter med t(11;14). Dette legemiddelet har foreløpig ikke MT/EMA-godkjenning for myelomatose, og er heller ikke vurdert i Nyemetoder. , men er et meget godt alternativ for disse pasientene. Dersom venetoclax vurderes brukt off label, er det Mmed dagens dokumentasjon er det naturlig å bruke venetoclax tidligst aktuelt i 4. linje, etter lokal godkjenning/vurdering fra Ekspertpanelet (Kumar et al., 2017; Kumar et al., 2020; Moreau et al., 2017).
Se også informasjon om behandling utenfor indikasjon:
- eHåndbok - Utprøvende behandling med legemiddel utenfor indikasjon («off-label»). (ous-hf.no)
- Utprøvende behandling - nasjonale prinsipper - Helsedirektoratet
Teclistamab og Cilta-Cel er har MT/EMA-godkjenning og under vurdering av beslutningsforum. Elranatamab og talquetamab er tilgjengelig som compassionate use. Melflufen og selinexor har begge MT/EMA-godkjenning, men er ikke markedsført i Norge.
- Ta hensyn til medikamenters bivirkningsprofil i lys av komorbiditet og vedvarende bivirkninger fra tidligere behandling
- Når pasienter er refraktære for enkeltmedikamenter, elimineres valgene fra listen nedenfor. Prioriteringene mellom dem er fortsatt den samme.
- Blant kombinasjoner på samme prioritering, velges det regime som gir endring av virkningsmekanisme fra det som har vært brukt, med størst fokus på det som ble brukt sist.
- Tripletter foretrekkes fremfor dubletter
- Listen er basert på medikamentkombinasjoner som har vært brukt i Norge.
- Revlimid og Velcade brukes med salgsnavn, mens for de andre bruker vi generiske navn. Dette fordi det er slik bruken av bokstavkombinasjoner har utviklet seg.
- OS-gevinst i fase-3-studier:
- Karfilzomib-Revlimid-Dexametason (KRd vs Rd) (Siegel et al., 2018)
- Elotuzumab-Revlimid-Dexametason (ERd vs Rd) (Dimopoulos et al., 2018b)
- Karfilzomib-Dexametason (Kd vs Vd) (Dimopoulos et al., 2018a)
- Daratumumab-Revlimid-Dexametason (DRd vs Rd)
- Elotuzumab-Pomalidomid-Dexametason (EPd vs Pd)
Må ha prøvd Revlimid
- PFS-gevinst:
- Daratumumab-Carfilzomib-Dexametason (DKd vs Kd)
- Isatuximab-Carfilzomib-Dexametason (IKd vs Kd)
- Isatuximab-Pomalidomid-Dexametason (IsaPd vs Pd)
- Daratumumab-Velcade-Dexametason (DVd vs Vd)
- Pomalidomid-Velcade-Dexametason (PVd vs Vd)
Må ha prøvd Revlimid - Ixazomib-Revlimid-Dexametason (IRd vs Rd)
- OS-gevinst over kun dexamethason i fase-3-studier:
- Revlimid-Dexametason (Rd vs D)
- Velcade-Dexametason (Vd vs D)
- Pomalidomid-Dexametason (Pd vs D)
Må ha prøvd Revlimid
- Fase-2-studier:
- Daratumumab (anbefaler å legge til Dexametason i vanlig dose)
- Daratumumab-Pomalidomid-Dexametason (Chari et al., 2017)
- Bendamustin-Velcade-Dexametason (Rodon et al., 2015)
- Bendamustin-Revlimid-Prednisolon (Beck et al., 2017)
- Bendamustin-Pomalidomid-Dexametason (Sivaraj et al., 2018)
- Venetoclax-Dex til pasienter med t(11;14) (Kumar et al., 2017)
- Cyklofosfamid-Velcade-Dexametason (de Waal et al., 2015)
- Cyklofosfamid-Revlimid-Prednisolon (Nijhof et al., 2016)
- Cyklofosfamid-Pomalidomid-Dexametason (Baz et al., 2016)
- Cyklofosfamid-Carfilzomib-Dexametason (Bringhen et al., 2014)
- Bendamustin-Carfilzomib-Dexametason
Primært refraktær sykdom
Vi anbefaler følgende håndtering i forbindelse med HMAS:
- For pasienter som har progresjon i starten av induksjonsbehandlingen => Bytt induksjonsbehandling, og involver en ekspert i valg av behandling.
- For pasienter som har progresjon sent i induksjonsbehandling => Gå raskt til HMAS
- For pasienter som ikke har progresjon, men suboptimal respons => Fullfør 4 sykluser induksjonsbehandling og gå raskt til HMAS
For pasienter som ikke skal til HMAS, behandler man videre til progresjon, og bytter så til det neste aktuelle behandlingsregimet.
Anbefalt behandling ved tilbakefall
HMAS kan være del av behandlingen hvis pasienten antas å tåle dette, og det er stamceller tilgjengelig. Man velger induksjonsbehandling på samme måte som ved tilbakefall, og kontinuerer behandlingen til progresjon etter HMAS
Anbefalt rekkefølge på regimerer
VRD => DVd => Kd => Pd => Belantamab. Se kommentarer til disse behandlingene tidligere i dokumentet.
Hvis pasienten ved første tilbakefall har nevropati og ikke kan få standard behandling med DVd, anbefales det behandling med daratumumab (eller isatuximab) i kombinasjon med carfilzomib-dex.
Hvis pasienten ikke er refraktær for lenalidomid, anbefales DRd ved første tilbakefall.
Mer detaljer om behandlingsregimer
K=Karfilzomib, R=Revlimid, d=dexametason, E=elotuzumab, D=daratumumab, V=Velcade, I=Ixazomib, Pan=panobinostat, P=pomalidomid, Benda=bendamustin, Pred=prednisolon, C=cyklofosfamid
Alle medikamenter som kan gis oralt eller subkutant, er dosert slik. Hvis ikke annet er beskrevet er behandlingen til progresjon eller manglende toleranse. Fordi pasienter har ulik behandlingshistorie, er det er beskrevet regimer her som ikke var nevnt i tabellen over
For alle behandlinger anbefaler vi ukentlig dexametason 40mg for pasienter under 75 år og ukentlig dexametason 20mg for pasienter over 75 år
Vd, Pd, KRd, Kd, ERd, EPd, IRd ogPVd
- Beskrevet i Felleskatalogen, med følgende unntak
- Dexametason iht beskrivelse ovenfor
- Carfilzomib gis alltid ukentlig, og i 70mg/m2, bortsett fra første dose som er 20mg/m2. I pasienter hvor man er usikker på toleransen er det lurt å trappe opp dosen via 36 og 56mg/m2.
Rd
- R som i Felleskatalogen
- Dexa ukentlig 40 mg peroralt
Dara (D)
- Beskrevet i felleskatalogen. Vi anbefaler tillegg av 40 mg dexametason ukentlig
DRd
- Daratumumab beskrevet i Felleskatalogen
- Rd som beskrevet ovenfor
DVd
- 28-dagerssykluser
- Daratumumab gis som beskrevet i Felleskatalogen for monoterapi
- Velcade dag 1,8 og 15 i de 8 første (4-ukers)-syklusene
- Dexametason 40 mg dag 1,8,15 i de 8 første (4-ukers)-syklusene
DPd
- 28-dagerssykluser
- Daratumumab som i monoterapi
- PomDex som i Felleskatalogen
IsaPd
- Isatuximab og PomDex, begge som i Felleskatalogen
DKd
- 28-dagerssykluser
- Daratumumab som i monoterapi
- Karfilzomib 20 mg/m2 dag 1 i syklus 1
- Karfilzomib 70 mg/m2 dag 8,15 i syklus 1 og dag 1,8,15 fra syklus 2
- Dexametason 40 mg ukentlig
BendaVd
- 28-dagerssykluser
- Bendamustin 70 mg/m2 dag 1 og 8
- Velcade 1,3 mg/m2 dag 1,8,15,22
- Dexametason 20 mg dag 1,8,15,22
- 6 sykluser hver 4. uke. Deretter 6 sykluser hver 8. uke. Til sammen 12 sykluser.
- Hvis god toleranse bør man vurdere å fortsette.
BendaRDexa
- 28-dagerssykluser
- Bendamustin 75 mg/m2 dag 1 og 2
- Revlimid 25 mg dag 1–21
- Dexametason 40mg ukentlig
- 8 sykluser
- Hvis god toleranse bør man vurdere å fortsette
BendaPd
- 28-dagerssykluser
- Bendamustin 120 mg/m2 dag 1 syklus 1–12
- Pomalidomid 3 mg dag 1–21 (fortsettes også etter syklus 12)
- Dexametason 40 mg dag 1,8,15,22 syklus 1–6
- Dexametason 20 mg dag 1,8,15,22 fra syklus 7
Venetoclax-Dex (kun aktuelt for pasienter med translokasjon 11;14)
- 21-dagerssykluser
- Venetoclax 800 mg 1–21
- Dexametason 40 mg ukentlig
- Pasienter >75 år halverer dosen dexametason
CVd
- Syklus 1–3: 21-dagerssykluser
- Velcade 1,3 mg/m2 dag 1,8
- Cyklofosfamid 50 mg dag 1–21
- Dexametason 40mg ukentlig
- Syklus 4–6: 35-dagerssykluser
- Velcade 1,6 mg/m2 dag 1,8,15,22
- Cyklofosfamid 50 mg dag 1–35
- Dexametason 40 mg ukentlig
- Syklus 7 og videre: 28-dagerssykluser
- Velcade 1,3 mg/m2 dag 1,15
- Cyklofosfamid 50 mg dag 1–28
CRDexa
- 28-dagerssykluser
- Revlimid 25 mg dag 1–21
- Cyklofosfamid 50 mg dag 1–28
- Dexametason 40mg ukentlig
CPd
- 28-dagerssykluser
- Pomalidomid 4 mg dag 1–21
- Dexametason 40 mg ukentlig
- Pasienter >75 år halverer dosen dexametason
- Cyklofosfamid 400 mg dag 1,8,15
CKd
- 28-dagerssykluser
- Cyklofosfamid 300 mg/m2 dag 1,8,15
- Karfilzomib 20 mg/m2 dag 1,2 i syklus 1
- Karfilzomib 36 mg/m2 dag 8,9,15,16 i syklus 1; dag 1,2,8,9,15,16 fra syklus 2
- Dexametason 40mg ukentlig
Benda-Kd
- 28-dagerssykluser
- Bendamustin 70 mg/m2 dag 1 og 8 syklus 1–8
- Karfilzomib 20 mg/m2 dag 1,2 i syklus 1
- Karfilzomib 27 mg/m2 dag 8,9,15,16 i syklus 1; dag 1,2,8,9,15,16 fra syklus 2–8
- Karfilzomib 27 mg/m2 dag 1,2,15,16 fra syklus 9
- Dexametason 40mg ukentlig
Spesielle forhold og bivirkninger ved nyere behandlinger
Belantamab:
Belantamab mafodotin er et nytt medikament med noen nye utfordringer. Medikamentet kan brukes fra 5.linje. Behandlingen er beskrevet i Felleskatalogen og gis i monoterapi. Vi vil likevel understreke noen ting å huske på:
- Behandlingen kan gi keratopati, og øyelegekontroll før oppstart og etter de 3 første syklusene er sterkt anbefalt. Det finnes retningslinjer for hva som skal vurderes av øyelege og hva som skal lede til utsettelse av behandling. Disse kan skaffes til veie av GSK. Man bør også snakke med pasientene om subjektiv synsreduksjon. Hvis det er tvil om man skal pausere behandling, kan man velge å la den subjektive synsreduksjonen styre. Bivirkningen er uansett reversibel. Sykdommens aggressivitet vil påvirke i hvilken grad man bør pausere behandlingen.
- Det anbefales å bruke en lavere frekvens av infusjoner helt fra starten. Vi anbefaler en startfrekvens på 4-6 uker, avhengig av aggressiviteten til sykdommen. Dette er altså lenger enn de 3 ukene som er definert i Felleskatalogen
Bispesifikke antistoffer og CAR-T:
Tre slike behandlinger er har MT/EMA-godkjenning og er markedsførte i Europa, men er ikke innført i Norge enda. Teclistamab, et bispesifikt antistoff mot BCMA, er vurdert med ikke godkjent av Nye Metoder (august 2023). Ide-cel/cilta-cel som er to CAR-T-produkter mot BCMA er til vurdering i Nye Metoder. I tillegg er elranatamab (bispesifikt antistoff mot BCMA) og talquetamab (bispesifikt antistoff mot GPRC5D) tilgjengelig på compassionate use, og flere nyere produkter eksisterer i kliniske studier i Norge. Det følgende er generell informasjon om håndtering av komplikasjoner relatert til disse behandlingene:
Cytokinstorm (cytokine release syndrome, CRS) er en feberreaksjon som kan utvikle seg til en alvorlig tilstand med multiorgansvikt. CRS skyldes aktivering av T-celler og makrofager. Diagnosen stilles klinisk og kan være vanskelig å skille fra infeksjon/sepsis. CRS oppstår typisk i løpet av den første uken etter CAR-T-celleinfusjon og innen 48 timer etter behandling med bispesifikke antistoffer (gjerne tidlig i første syklus). Sent-innsettende CRS er sjelden, men kan oppstå f.eks. hvis bispesifikke antistoffer gis i forbindelse med (lavgradig) infeksjon eller etter opphold i behandlingen.
CRS behandles etter alvorlighetsgrad. I tillegg til nødvendig støttebehandling, gis tocilizumab (IL-6 reseptorhemmer) og kortikosteroider. Avdelinger som behandler slike pasienter må ha tocilizumab og en algoritme for behandling av CRS tilgjengelig.
Nevrotoksisitet er mindre hyppig og forekommer typisk under eller etter CRS. Tilstanden kan gi nedsatt konsentrasjon og forvirring. Alvorlige former kan gi redusert bevissthet og hjerneødem. Det er også beskrevet andre typer nevrologiske bivirkninger som kan oppstå senere.
Cytopenier og infeksjoner er vanlige komplikasjoner. Cytopeniene kan være langvarige. Behandlingen rettes også mot normale B-celler og plasmaceller og hypogammaglobulinemi er vanlig. Pasientene kan også ha T-celledefekter med risiko for reaktivering av CMV og EBV samt økt risiko for alvorlige forløp av COVID-19, influensa, RS-virus, parvovirus, adenovirus og andre virale infeksjoner. Erfaringene så langt tilsier at denne risikoen er større ved bispesifikke antistoffer enn ved CAR-T-celler.
- Immunglobulinsubstitusjon anbefales ved IgG < 4 g/L.
- G-CSF kan gis, men bør unngås i den første fasen pga risiko for forverring av CRS.
- Herpes zoster profylakse (Valaciclovir 250 mg x2) og pneumocystis jiroveci profylakse (f.eks. Bactrim 1 tablett daglig eller hver annen dag) bør gis. Anbefalt varighet etter CAR-T-celleinfusjon er minst 1 år og til CD4 T-celler er over 200/µL. Ved bispesifikke antistoffer bør profylaksen trolig beholdes til etter avsluttet behandling.
Tromboseprofylakse
Sist faglig oppdatert: 21.12.2023
Tromboseprofylakse ved thalidomid, lenalidomid og pomalidomid:
- Acetylsalicylsyre 75 mg. Hvis pasienten har høy tromboserisiko bør warfarin, DOAC eller lavmolekylært heparin (enoxaparin 40 mg eller ekvivalent) brukes.
Veiledende dosejusteringer ved benmargs- og nyresvikt
Sist faglig oppdatert: 21.12.2023
Pegylert G-CSF bør alltid prøves for å opprettholde doseintensitet.
Ved nyresvikt
Melfalan 25–50 % reduksjon ved GFR 30–50 ml/min, 50–75 % reduksjon ved eGFR<30 ml/min.
Cyklofosfamid er upresist rapportert i litteraturen, 25–50 % reduksjon er anbefalt ved 25–75 % reduksjon av clearance.
Bortezomib, bendamustin, carfilzomib, daratumumab og pomalidomid kan gis i full dose ved nyresvikt, men det er mindre erfaring ved alvorlig nyresvikt (GFR<10 ml/min).
Revlimid kan startes i 15 mg ved eGFR <30 ml/min, men man må følge celletall og kreatinin ukentlig i startfasen.
Strålebehandling
Sist faglig oppdatert: 21.12.2023
Myelomatose er vanligvis en strålefølsom sykdom. Ved solitære plasmacytomer kan strålebehandling alene være kurativt. Sykdommen er imidlertid ofte generalisert og systemisk behandling er da å foretrekke som hovedbehandling. Strålebehandling kan være nyttig for kontroll av lokale problemer som smertefulle lesjoner og truende frakturer.
Det oppstår ofte spørsmål om rekkefølgen hvis man skal gi flere behandlingsmodaliteter. Generelt anbefales å prioritere medikamentell behandling for å spare benmargsreserver. Behovet for stråling vil ofte falle bort hvis kjemoterapi gis først.
Kan strålebehandling og kjemoterapi gis samtidig?
For eldre medikamenter (alkylerende agens, antracykliner, cisplatin) er det nødvendig å ha minst 4 ukers intervall mellom kjemoterapi og strålebehandling. Dokumentasjon mangler for de nye medikamentene, men erfaringen er at dette kan gis samtidig. Vi mener derfor at dette kan prøves hvis det ansees tidsmessig nødvendig.
Rebestråling
Mange pasienter vil trenge strålebehandling gjentatte ganger i sitt sykdomsforløp. Begrensende for rebestråling vil være tilgrensende normalvevs toleranse. Dette vurderes av stråleenheten. Hvis effekten tidligere har vært god og varig (>6–12 måneder), vil det som regel være trygt og sannsynligvis nyttig å gi ny bestråling.
Tilbakefall med plasmacytomer
Hvis pasienten får et nytt plasmacytom under behandling der det er god kontroll på den systemiske sykdommen, anbefales stråling og kontinuering av den systemiske behandlingen.
Hvis pasienten får et nytt plasmacytom i behandlingspause, anbefales restart av systemisk behandling, og vurdering av om stråling trengs i tillegg.
Kirurgisk behandling
Kirurgisk behandling av myelomatosepasienter er aktuelt ved fraktur, truende fraktur og sammenfall i columna med truende tverrsnittslesjon.
Det er ingen kontrollerte randomiserte kliniske studier angående kirurgisk behandling av skjelettmetastaser. Resultatene etter operativ behandling fremkommer i retrospektive materialer som omfatter flere kreftformer. Brystkreftmetastaser dominerer i slike materialer, og myelomatose utgjør bare 10–15 %. Metastaser til skjelettet opptrer og forløper forskjellig for forskjellige krefttyper, og det er funnet bedre overlevelse for myelomatosepasienter som er operert i skjelettet enn for andre. Det er alt i alt sparsomt med systematisk informasjon, og beslutningene for myelomatosepasienter må i hovedsak tas på et generelt erfaringsgrunnlag. Kirurgisk behandling bør tilstrebe å gi rask mulighet for rehabilitering, liten risiko for alvorlige komplikasjoner og et resultat som ikke forventes å kreve reoperasjon i pasientens levetid.
Kyfoplastikk, hvor man injiserer sement i en frakturert vertebra, er også aktuelt, og kan i en del tilfeller ha god smertelindrende effekt.
Situasjoner hvor strålebehandling, kirurgisk behandling og kjemoterapi vurderes opp mot hverandre
Sist faglig oppdatert: 21.12.2023
Truende tverrsnittslesjon
Kompresjon av medulla spinalis opptrer hos 5–10 % av pasienter med myelomatose og er en medisinsk akuttsituasjon. Slike manifestasjoner skyldes osteolytiske destruksjoner med sammenfall av vertebrae eller en bløtdelstumor som komprimerer medulla.
Tverrsnittslesjon skal mistenkes ved:
- Nytilkomne ryggsmerter (ofte belteformete)
- Nevrologiske symptomer fra underekstremitetene (både sensoriske og motoriske), nedsatt kraft, gangproblemer
- Sfinkter- eller tarmparese med inkontinens, obstipasjon/urinretensjon (sene tegn)
Funn ved klinisk undersøkelse: Slapp analsfinkter, paraparese, sensibilitetstap på underekstremiteter, sensibilitetsforskjell på trunkus.
Pasienten bør startes raskt på steroider som har god ødemdempende virkning og bør få strålebehandling innen 1 døgn. Steroiddose dexametason 40 mg daglig i opptil 4 dager.
Smertefulle lesjoner
Kjemoterapi eller strålebehandling er aktuelt. Dersom pasienten er vanskelig å smertelindre med kjemoterapi eller analgetika, er det god indikasjon for strålebehandling. Man har tradisjonelt gitt 20–30 Gy over 1–2 uker, men man kommer som regel til målet med enkel fraksjonering 8 Gy x 1. En slik dose kan godt gjentas mot samme område.
Truende fraktur og fraktur
Kirurgi/strålebehandling er aktuelt. Det er utarbeidet kriterier for kritisk skjelettstyrke, men disse blir i praksis lite brukt. Indikasjonen bygger på en klinisk vurdering hvor man legger vekt på smerter, funksjon, generell helsetilstand og forventet overlevelse.
Pasient med store smerter som øker ved belastning og påvist lesjon med fortynning av corticalis er kandidat for profylaktisk behandling. Slike pasienter bør diskuteres med ortoped og onkolog. Ved antatt stor frakturfare er det rimelig å velge profylaktisk kirurgi. Det finnes ikke studier som viser at strålebehandling forebygger fraktur. I overarmsben og i rygg vil det sjelden være indikasjon for operasjon før fraktur har oppstått.
Fraktur bør behandles kirurgisk hvis ortopedisk indikasjon.
Postoperativ strålebehandling
Frakturkirurgi vil sjelden eliminere tumor og det kan øke mulighetene for et godt langtidsresultat å gi postoperativ strålebehandling. Det er derfor vanlig å gi 3 Gy x 10 i denne situasjonen. Selv om dokumentasjonen er svak er det rimelig å strålebehandle frakturer som ikke opereres.
Andre romoppfyllende prosesser som truer viktige strukturer
Eksempler på slike situasjoner er bløtdelstumores med avklemming av galle- eller urinveier, kompresjon av hjernenerver, protrusjon av orbita. Strålebehandling med 3 Gy x 10 vil oftest være førstevalg, men stenting kan være aktuelt i galle- og urinveier.
Solitært plasmacytom i ben
Solitære plasmacytomer utgjør 5 % av alle plasmacelletumores. Det foreligger som regel en lytisk solitær destruksjon av ben, og biopsi gir diagnosen. Det må samtidig gjøres utredning med tanke på om det foreligger flere plasmacytomer eller generalisert sykdom. Det viktigste er å identifisere eventuelle andre plasmacytomer da tilstanden fortsatt er potensielt kurativ. Pasientene kan ha M-komponent av varierende størrelse, og denne kan skyldes plasmacytomet, særlig hvis den blir borte etter stråling. Erfaringen viser at 75 % av pasientene utvikler myelomatose i løpet av 2–4 år. I utgangspunktet skal man likevel ha kurativ målsetting, også i tvilstilfeller. Dette har først og fremst betydning for dosering av strålebehandlingen. Supplerende systemisk behandling er ikke indisert ved asymptomatisk sykdom. Hvis man er engstelig for at det foreligger en systemisk restsykdom, kan denne uansett ikke helbredes ved systemisk behandling.
Ved kurativt siktemål gis høyere doser og fraksjonering, anbefalt dose er 2 Gy x 20 og ved plasmacytomer >5 cm vurderes gitt 2 Gy x 25.
Ekstramedullært plasmacytom
Disse utgjør 3–5 % av plasmacelleneoplasiene og 80 % finnes i øvre respirasjonstraktus eller ØNH-området. Disse behandles som de andre plasmacytomene, selv om man kan diskutere nytten av strålebehandling i de tilfellene der tumor er kirurgisk fjernet i sin helhet. Ved tvil bør strålebehandling gis i kurative doser. Ca. 30 % av pasienter med ekstramedullære plasmacytomer vil senere utvikle systemsykdom.
Anbefalinger plasmacytomer (i ben eller ekstramedullært)
Diagnostikk: vanlig myelomatoseutredning pluss MR av columna og bekken for å påvise eventuelle andre lesjoner. PET-CT ved ekstramedullært plasmacytom. For oppfølging, se tidligere avsnitt.
Behandling: ved ekte solitært myelom gis strålebehandling med kurativ dose. Ved påvisning av flere lesjoner eller myelomatose gis primært systemisk behandling som ved myelomatose.
Truende tverrsnittslesjon:
Diagnostikk: MR columna rekvireres som ø.hj., evt CT myelografi dersom kontraindikasjon mot MR.
Behandling: Dexametason 40 mg x1 startes umiddelbart.
Pasienten overføres til sykehus med strålebehandlingstilbud, og helst nevrokirurgisk avdeling. Strålebehandling 3 Gy x10 startes innen 24 timer. Hvis progressive symptomer, behov for stabilisering, usikker diagnose eller hvis strålebehandling ikke kan startes, vurderes akutt laminectomi.
Støttebehandling
Sist faglig oppdatert: 21.12.2023
Bisfosfonater og denosumab
Behandling med bisfosfonater reduserer skjelettskade og gir økt totaloverlevelse. Også pasienter uten skjelettskade ved diagnosetidspunkt har effekt. Både bisfosfonater og denosumab kan gi osteonekrose i kjeven etter langvarig behandling. Pasienter bør vurderes av tannlege før oppstart av bisfosfonat, og alle pasienter bør få tillegg av Kalsiumpreparat med D-vitamin. dosering 1000/800.
Anbefaling profylaktisk behandling med bisfosfonater:
- Zoledronat 4 mg iv. over 15 min hver 4. uke. Behandlingstid 2 år. Hvis pasienten oppnår VGPR, kan behandling avsluttes etter minst 1 års behandling.
Ved tilbakefall må bisfosfanatbehandling vurderes i den enkelte pasient. Man må vurdere totalmengden bisfosfanat de har fått, hvorvidt de har skjelettsykdom og hvorvidt den er økende. Hvis man starter behandling på nytt kan den begrenses til ett års behandling, og man kan vurdere å øke intervallet opp mot 3 måneder. Bisfosfonater anbefales ikke hvis GFR<30 ml/min. Denosumab kan brukes i denne situasjonen, men man må være obs på potensiell hypokalsemi. Det skal gis en avsluttende dose zoledronsyre når man avslutter denosumab.
Anbefaling infeksjonsprofylakse
- Ved kombinasjonen hypogammaglobulinemi og residiverende infeksjoner med kapselkledde bakterier anbefales substitusjon med immunglobulin. Det samme med kombinasjon av hypogammaglobulinemi og én alvorlig infeksjon med kapselkledde bakterier. Ved stor infeksjonsproblematikk som ikke fyller disse kriteriene, kan det også forsøkes. Det brukes forskjellige preparater i forskjellige helseforetak, se egne instrukser for dosering. Behandlingen avsluttes hvis infeksjonstendensen ikke bedres. Pasienter som behandles med bispesifikke antistoffer skal alltid substitusjon med immunglobuliner hvis IgG faller under 4.
- Anbefaling Herpes Zoster profylakse ved proteasomhemmer, CD-38 antistoff og til 3 måneder etter HMAS.
- Valtrex 250 mg x 2 po, avsluttes 1–2 måneder etter proteasomhemmer, og 3 mnd etter CD-38-antistoff.Vi anbefaler den rekombinante herpes-zoster-vaksinen til alle pasienter etter HMAS, og også til alle andre myelomatosepasienter.
Anemibehandling
Ved anemi er det viktig å utelukke andre vanlige årsaker. Et minimum er utredning med retikulocytter, ferritin, B12, folat og homocystein. Hvis ingen andre årsaker kan påvises og det er behov for behandling av anemi, kan erytropoiesestimulerende medikamenter (ESA) prøves.
Behandling av hypercalcemi
Sist faglig oppdatert: 21.12.2023
Anbefaling:
Anbefaling behandling av hyperkalsemi:
- Moderat/alvorlig hyperkalsemi (s-Ca>2,90 mmol/l eller ionisert Ca>1,45):
Rehydrering, 4–6 l NaCl. iv., diuretika etter behov. - Bisfosfonat dosering som anbefalt i Legemiddelhåndbok, effekt etter 1–2 døgn og max effekt etter 3–7 døgn.
- Steroider som ved myelomatosebehandling, effekt innen 3–4 dager.
- Myelomatosebehandling startes raskt.
- Iv Calcitonin hvis rask reduksjon av Ca ønskes, må gjentas pga kort halveringstid.
Vurdering av behandlingsrespons og kontroll
Sist faglig oppdatert: 21.12.2023
Hensikten er å registrere beste behandlingsrespons etter behandling og deretter å oppdage tilbakefall. Vurdering gjøres i forbindelse med hver syklusstart. Kan forlenges til 2–3 sykluser ved vedlikeholdsbehandling i førstelinje. Pasienten følges med samme blod- og urinprøver som ved diagnose, bortsett fra b2-mikroglobulin.
Pasienter med oligo- og nonsekretorisk sykdom må følges med benmarg og PET-undersøkelser. I førstelinje følges pasienten hver 6. måned med biopsi og PET annenhver gang. Fra første tilbakefall følges pasienten hver 3. måned med biopsi og PET annenhver gang.
Billedfremstilling
Det bør alltid gjøres lavdose CT ved hver behandlingsstart, for å kunne påvise progresjon ved bilder tatt ved klinisk indikasjon, men ikke hyppigere enn hver 6. måned. Forøvrig tas bilder bare ved klinisk indikasjon.
Responskriterier (Kumar et al., 2016)
Responskriterier er utviklet for bruk i studier og for sammenligning mellom forskjellige institusjoner, men er også nyttige ved oppfølging i rutinepraksis. Minor respons (MR, >25 % reduksjon av M-protein) brukes bare ved tilbakefall. Skillet mellom CR og sCR har bare betydning i studier og systematisk oppfølging.
For å kunne evaluere respons kreves det at det er målbar sykdom i den kategorien prøver man evaluerer. Denne grensen er 10 g/l for s-m-komponent, 200 mg/døgn for u-m-komponent i døgnurin, og 100 mg/l for involvert lettkjede. Lettkjedemåling kan bare brukes hvis det ikke er målbar sykdom for s-m-komponent eller u-m-komponent. Hvis det ikke er gjort døgnurin, er det mest praktiske å vurdere denne som «ikke målbar».
Responskategori | Definisjon av responskategori |
|
|---|---|---|
MRD negativ | MRD negativitet målt ved flowcytometri (EuroFlow) eller next generation sequencing (NGS) |
|
s (stringent) CR | CR, i tillegg normal S-FLC ratio og ingen klonale plasmaceller i benmarg ved immunhistokjemi (biopsi) eller immunfluorescens (flow) |
|
CR (complete response) | M-komponent 0, negativ immunfiksasjon på serum og urinelektroforese, regress av bløtdelsplasmacytomer, ≤5 % plasmaceller i benmarg. Hvis pasienten følges med lettkjedemålinger: normal lettkjederatio. |
|
VGPR (very good partial response) | ≥90 % reduksjon av serum M-komponent og urin M-komponent <100 mg/ 24 timer eller serum og urin M-komponent kan ikke påvises ved elektroforese, men ved immunfiksasjon. Hvis ikke målbar M-komponent i serum eller urin, reduksjon av differanse mellom involvert og ikke-involvert lettkjeder (dFLC) >90 %. Ekstramedullære plasmacytomer skal reduseres med ≥90 % |
|
PR (partial response) | ≥50 % reduksjon av serum M-komponent og reduksjon av 24 timers urin M-komponent med ≥90 % eller <200 mg/24 timer. Hvis ikke målbar M komponent i serum eller urin, reduksjon av dFLC >50 %. Hvis serum og urin M-komponent ikke er målbar, og serum FLC også er normal, kreves det ≥50 % reduksjon av antall plasmaceller i benmargen, forutsatt at baseline plasmacelleandel var ≥30 %. Ekstramedullære plasmacytomer skal reduseres ≥50 % i størrelse |
|
MR (minimal response) | ≥25% reduksjon av serum M-komponent og reduksjon av 24-timers urin M-komponent med >50% Hvis ikke målbar M-komponent i serum eller urin, reduksjon av dFLC >25% Ekstramedullære plasmacytomer skal reduseres ≥50 % i størrelse | |
SD (stable disease) | Tilfredsstiller ikke kriterier for sCR, CR, VGPR, PR eller PD |
|
PD (progressive disease) | Stigning av M-komponent på ≥25 % fra baseline (men minst 5 g/l) og/eller urin M-komponent (minst ≥200 mg/24 timer). Hos pasienter uten målbar M komponent i serum eller urin: ≥25 % stigning av dFLC (absolutt stigning skal være >100 mg/L). ≥25 % økning av prosentandel plasmaceller i benmarg (men skal være minst 10 %) Utvikling av nye CRAB kriterier, se omtale i avsnitt Diagnostiske kriterier |
|
«Minimal residual disease» (MRD)
Sist faglig oppdatert: 21.12.2023
MRD kan defineres som restsykdom som kun kan detekteres med de mest sensitive metoder som er tilgjengelig. De senere år har det blitt utviklet mer sensitive undersøkelser av benmargen, flowcytometri, DNA sekvensering og PET-CT. I det første tilfellet har det vært utarbeidet en spesiell kombinasjon av markører som har vært benevnt «euroflow» og «Next generation flow» (NGF). Denne undersøkelsen kan under ideelle betingelser detektere en kreftcelle blant 105 benmargsceller. DNA sekvensering identifiserer kreftspesifikke mutasjoner og har en sensitivitet som er ca. 10x bedre. Myelomatose har flere fasetter og vokser også som plasmacytomer. PET-CT og MR kan oppdage små ansamlinger av myelomceller som ikke nødvendigvis detekteres av flow eller sekvensering av benmargsceller. Det er 10–15 % diskordans mellom de forskjellige MRD-metodene. MRD-analyser og den behandlingsmessige betydningen av disse er under rask utvikling. Det er tydelig vist at pasienter som blir MRD negative har en bedre prognose enn de som ikke oppnår dette, men vi snakker fortsatt ikke om kurasjon. Per i dag er det uklart hvorledes MRD-analyser bør brukes i praksis. Vi anser MRD ved flow, sekvensering eller PET-CT som et relevant endepunkt i kliniske studier. I vanlig klinisk praksis utenom studier er det imidlertid fullgodt å bruke de tradisjonelle responskriteriene. MRD kan også være aktuelt for kvalitetssikring lokalt.
Al-amyloidose
Sist faglig oppdatert: 23.12.2021
Anbefalinger:
- Ved lokalisert amyloidose anbefales kirurgisk eksisjon av affisert område hvis dette er klinisk indisert. Hos pasienter hvor eksisjon ikke er mulig er behandling med laser eller radioterapi en mulighet.
- For pasienter under 70 år uten komorbiditet er høydosebehandling med autolog stamcellestøtte (HMAS) førstelinjebehandling.
- Induksjonsbehandling med daratumumab- CyBorDex anbefalt til alle som skal til HMAS.
- Der daratumumab-CyBorDex ikke kan benyttes, anbefales CyBorDex eller MelBorDex.
- Førstelinjebehandling til pasienter som ikke er kandidat for HMAS er avhengig av Mayo stadium.
- For pasienter i Mayo stadium I anbefales daratumumab-CyBorDex.
- For pasienter i Mayo stadium 2 og 3A anbefales dosejustert daratumumab-CyBorDex.
- For pasienter i Mayo stadium 3B anbefales behandling med doseredusert MelBorDex.
Introduksjon og epidemiologi
Sist faglig oppdatert: 23.12.2021
Amyloidose er et samlebegrep på en rekke ulike tilstander som alle er karakterisert ved proteinavleiringer i ett eller flere organer. Proteinavleiringene betegnes amyloid og består av feilfoldede proteiner anordnet som fibriller. Farget med kongorød fremstår alle former for amyloid som rødt amorft materiale under vanlig lys og lysegrønt/eplegrønt under polarisert lys. Avhengig av hvilket protein som er avleiret finnes det en rekke forskjellige typer amyloidose, hvor AL amyloidose er den hyppigste systemiske varianten.
Ved AL amyloidose består avleiringene hovedsakelig av en klonal lettkjede som produseres av en klonal B-cellepopulasjon. Alle pasienter med systemisk AL amyloidose har en underliggende plasmacelle eller B-celle klon.
Systemisk AL amyloidose er en relativt sjelden sykdom. Insidensen i Norge er ikke kjent, men antatt insidens er 1 per 100 000/år (Kyle et al., 1992; Nienhuis et al., 2016). Det er betydelig overlapp mellom amyloidose og myelomatose og 5–10 % av pasienter med myelomatose har amyloidose. Amyloidose kan også forekomme ved en rekke andre klonale B-celle sykdommer som KLL og Waldenströms makroglobulinemi, men forekomst av amyloidose ved disse tilstandene er under 1 %.
I tillegg til AL amyloidose eksisterer det flere enn 30 andre proteiner som kan avleire seg som amyloid (transthyretin (TTT), serum amyloid (SAA) etc.). For behandling av disse formene vises til «veileder for diagnostikk og behandling av amyloidose». Mens noen av disse tilstandene er ervervet, har man de senere årene blitt klar over at det finnes mange arvelige former for amyloidose. Andre former for amyloidose har overlappende kliniske manifestasjoner, men skal ha annen oppfølgning og behandling enn AL amyloidose. Det har tidligere vært problem med å skille de ulike formene for amyloidose fra hverandre, men forbedret diagnostikk med massespektrometri gjør det nå mulig.
Amyloid kan forekomme i alle organer, men affeksjon av hjerte, nyre, nervesystem og gastrointestinale organer (lever og tarm) er hyppigst. Organinfiltrasjon fører til organdisfunksjon og end- stage organskade hvor restriktiv kardiomyopati og nefrotisk syndrom er de vanligste. Affeksjon av hjerte er også den viktigste prediktor av morbiditet og mortalitet ved AL amyloidose
Makroglossi, koagulasjonsfaktor X mangel og negledystrofi er typiske funn ved AL amyloidose, og periorbitale ekkymoser («racoon eyes») er patognomoniske for AL amyloidose. Disse funnene er kun til stede hos et fåtall av pasientene.
Lokalisert amyloidose
Sist faglig oppdatert: 23.12.2021
Hos noen pasienter påvises kun AL-amyloid avleiringer i ett organ mens det ikke kan påvises noen M-komponent i serum/urin, aberrant kappa/lambda ratio eller monoklonal B-celleklon. Disse tilfellene klassifiseres som lokalisert amyloidose. Lokalisert amyloidose forekommer oftest i luftveier, orbita, gastrointestinal trakt, lymfeknuter og hud.
Det er viktig å skille lokalisert fra systemisk AL amyloidose da behandling og prognose er forskjellig. Fullstendig undersøkelse som beskrevet i neste avsnitt er påkrevd for å avkrefte systemisk amyloidose.
Behandling er bare aktuelt hvis det er et klinisk behov. Ved behandling tilstrebes total kirurgisk eksisjon av affisert område. I de organer hvor kirurgisk eksisjon er vanskelig, eks. blære eller luftveier, vil laser og/eller ekstern strålebehandling være alternative behandlingsmuligheter. Systemisk terapi er kun indisert der det ikke er mulig å gjennomføre lokal behandling.
Diagnostikk og screening
Sist faglig oppdatert: 23.12.2021
Utredning ved amyloidose har følgende mål:
- Påvise og korrekt klassifisere amyloid.
- Verifisere at det foreligger systemisk sykdom.
- Kartlegge utbredelse av organdysfunksjon.
- Ta stilling til om det foreligger samtidig myelomatose eller annen klonal B-celle sykdom.
For å kunne stille diagnosen systemisk AL amyloidose, må alle av følgende fire kriterier være oppfylt:
- Histologisk verifiserte amyloidavleiringer med positiv kongorød farging eller amyloid fibriller ved elektron mikroskopi
- Verifisering av AL subtype (samt utelukke andre amyloidformer)- dette gjøres enten ved bruk av immunoelektronmikroskopi eller massespektrometri
- Påvist monoklonalt plasma celle proliferasjon ved en eller flere av følgende:
- M-komponent i serum ved immunfiksering
- M-komponent urin ved immunfiksering
- Aberrant kappa/ lambda ratio.
- Klonale plasmaceller i biopsi
- Påvist amyloidoserelatert organskade
Benmargsbiopsi sammen med biopsi av subkutant fett påvise amyloid hos 85 % pasienter. Der disse er negative, og biopsi av affisert organ er betenkelig (f.eks. blødningsrisiko) utføres først spyttkjertel og/eller rectum biopsi. Hvis disse biopsiene er negative, og mistanke om amyloidose er fortsatt høy, gjennomføres biopsi av affisert organ.
Ved biopsi av subkutant fettvev må man sørge for at biopsien har tilstrekkelig størrelse, dvs 1 cm3. Stansebiopsi fra hud er ikke tilstrekkelig så fremt det ikke foreligger kliniske tegn til amyloidose i hud. Alternativ til biopsi av subkutant fettvev er aspirasjon av subkutant fettvev med stor sprøyte og tykk nål; såkalt «fat-pad» aspirasjon. Imidlertid har «fat-pad» aspirasjon høy andel falsk negative prøvesvar hvis man ikke behersker denne teknikken tilstrekkelig. Biopsi av subkutant fettvev er derfor å foretrekke.
Immunhistokjemi anvendes primært for verifisering av AL amyloidose samt for å utelukke andre ulike typer amyloid. Immunhistokjemi er tilgjengelig ved de aller fleste avdelingene for patologi. Imidlertid vil man ved immunhistokjemi i en relativt høy andel av tilfellene ikke klare eller feil klassifisere hvilken type amyloid-avleiring som foreligger. Spesifisitet og sensitivitet avhenger av biopsisted og erfaring.
Massespektrometri av biopsimateriale vil i nesten alle tilfeller korrekt klassifisere amyloid-avleiringene. Metoden tilbys som rutineundersøkelse ved Oslo Universitetssykehus. Biopsier skal sendes til verifisering med massespektrometri ved avdeling for patologi OUS i alle tilfeller hvor konvensjonell histologi/immunhistologi har påvist eller gitt mistanke om amyloidose (uavhengig av type) slik at feilklassifisering og feilbehandling unngås.
Evaluering av klonal plasmacellesykdom
Sist faglig oppdatert: 23.12.2021
De aller fleste pasienter med AL amyloidose har målbar M- protein eller S- frie lette kjeder selv om absolutte verdier ofte er lavere en ved myelomatose.
Amyloidavleiringer i benmargen kan påvises hos ca. 30 % nydiagnostierte myelomatose pasienter uten at det foreligger amyloidrelatert organskade mens ca. 15 % av disse kan utvikle symptomatisk amyloidose.
Det er svært sjeldent at pasienter med AL amyloidose utvikler symptomatisk myelomatose.
Benmargsundersøkelse med biopsi og cytogenetiske analyser anbefales hos alle pasienter
Cytogenetiske avvik kan påvises hos opptil 90 % pasienter hvor t (11;14) er den vanligste, og forekommer hos 50–60 %. Andre relativt vanlige avvik er kromosom 13 avvik (monosomi 13, 13q del), trisomier og gain 1q.Del 17 p er sjelden.
Følgende analyser skal gjennomføres hos alle pasienter:
- Proteinelektroforese med immunfiksasjon
- Serum frie lette kjeder
- Urinproteinelektroforese med immunfiksasjon
Disse analyser vil kunne påvise klonal plasmacellesykdom i >98 % av tilfeller.
Kriterier for monoklonal plasmacelleproliferasjon (M komponent i serum eller urin, patologisk lettkjede ratio eller klonale plasmaceller) er ikke til stede hos ca. 2 % av pasienter med systemisk AL amyloidose. I disse tilfeller stilles diagnosen med forsiktighet etter nøye vurdering av histologiske funn og organsymptomer.
Samtlige pasienter med AL amyloidose og plasmacelleandel ≥10 % skal også evalueres med henblikk på symptomatisk myelomatose, for informasjon vedrørende utredning henvises til kapittelet om Myelomatose.
Spesielle situasjoner
- Funn av restriktiv kardiomyopati eller fortykket septum ved ekko cor uten andre holdepunkter for amyloidose enn M-komponent eller økte frie letter kjeder.
Diagnostikk av hjerteaffeksjon ved AL amyloidose baserer seg på enten påvising av amyloid i biopsier fra ekstrakardialt organ (eks. benmarg, eller fettvev), M‑komponent/frie letter kjeder urin og forhøyet NT-pro-BNP. Hvis man ikke kan påvise amyloid i biopsi fra ekstrakardielle organer og det fortsatt er mistanke om hjerteamyloidose anbefales direkte biopsi av hjertet.
Hos pasienter hvor man mistenker andre former for amyloidose kan man vurdere MR hjerte eller 99mTc-DPD scintigrafi før hjertebiopsi. 99mTc-DPD scintigrafi er tilgjengelig ved SUS, St. Olavs Hospital og OUS. Pasienter med AL amyloidose vil ikke ha opptak av tracer i hjertet, mens man ved senil amyloidose og TTR amyloidose har opptak av tracer i myocard.
- Isolert polynevropati.
Hos noen pasienter kan man påvise M-komponent sammen med polynevropati uten at man kan finne annen organmanifestasjon. Disse pasienter skal henvises til utredning hos nevrolog for å utelukke andre ervervede eller hereditære former for polynevropati. Biopsi av perifere nerver er frarådet da det sjelden gir bedre diagnostikk og gir varige men. Det må gjennomføres biopsi fra benmarg og subkutant fett.
IgM amyloidose og amyloidose ved andre lymfoproliferative tilstander
Sist faglig oppdatert: 23.12.2021
IgM amyloidose utgjør ca. 5 % av alle tilfeller med AL amyloidose. Denne har spesifikk klinisk presentasjon med høyere forekomst av affeksjon av lunger, lymfoknuter og perifert nervesystem. Det er utarbeidet en egen prognostisk model for IgM AL amyloidose basert på kardielle biomarkører, affeksjon av lever og perifert nervesystem. Pasienter uten risikomarkører har medianoverlevelse på 90 mnd sammenlignet med 33 mnd ved tilstedeværelse av 1 risikofaktor og 16 mnd ved kombinasjon av to eller flere faktorer (Sachchithanantham et al., 2016).
AL amyloidose kan også forekomme ved andre lymfoproliferative tilstander. I disse tilfellene er det viktig å utelukke at det foreligger en liten klonal plasmacellepopulasjon. Hvis det ikke foreligger en annen plasmacellepopulasjon startes behandling mot lymfoproliferativ tilstand med mål om komplett respons.
Evaluering av organaffeksjon etter verifisert AL amyloidose
Sist faglig oppdatert: 23.12.2021
Hos alle pasienter med verifisert AL amyloidose må man kartlegge organaffeksjon og grad av denne. Anbefalt utredning av organaffeksjon er oppsummert i tabell 9.1.
Tabell 9.1
Organ | Utredning |
|---|---|
Hjerte | Alle pasienter: Troponin og NT-Pro-BNP EKG (low voltage) Ekkokardiografi (systolisk og diastolisk funksjon, intraventrikulær septum fortykkelse) 24-timers telemetri/Holtermonitorering anbefales til alle med påvist hjerteamyloidose og ved klinisk mistanke om arytmi. |
Nyre | Alle pasienter: Kreatinin, eGFR, s-albumin U- albumin/ kreatinin ratio |
Lever | Alle pasienter: Leverfunksjonstester spes ALP Evaluering av leverstørrelse (klinisk eller billeddiagnostisk) |
Tarm | Ved indikasjon Gastro-og/ eller koloskopi med biopsier ved gastrointestinale symptomer inkl. blødning |
Nevropati | Alle pasienter: Klinisk evaluering Ortostatisk BT måling Ved klinisk indikasjon Elektromyografi og nevrografi |
Koagulasjon | Alle pasienter: INR, APTT. Faktor X måling hvis INR er over referanseområde eller ved klinisk blødningstendens |
Endokrinopati | Alle pasienter: Thyroidea funksjonstester, vitamin D og fastende blodsukker Ved indikasjon kjønnshormoner |
Lunge | Ved indikasjon Spirometri og røntgen thorax |
Screening mtp amyloidose hos pasienter med MGUS/myelomatose
Sist faglig oppdatert: 23.12.2021
Alle pasienter med MGUS og personer med nydiagnostisert myelomatose skal undersøkes og følges med:
- Albumin/Kreatinin ratio i urin.
- NT-pro-BNP.
Prognose og stadieinndeling
Sist faglig oppdatert: 23.12.2021
Prognosen ved AL amyloidose predikeres hovedsakelig av følgende to faktorer:
- Hjerteaffeksjon: vurderes ved hjelp av kardielle markører som pro BNP (eller BNP) og Troponin T (eller troponin I)
- Differansen mellom involverte og ikke-involverte FLC (dFLC).
Det er utarbeidet flere forskjellige prognostiske modeller. Mest brukte er Mayo-stage-inndeling. På grunnlag av denne er det senere utarbeidet to andre systemer, Mayo-stage (2004/) European og revised-Mayo-stage. Det europeiske systemet benytter svært høye NT-proBNP nivåer for å identifisere høyrisiko pasienter. Revised Mayo benytter differansen mellom involverte og ikke involvert FLC (dFLC) sammen med NT-proBNP og troponin. Mayo2004/European har vist å ha best prediktiv verdi for identifisering av pasienter med lav–risiko og svært høy risiko.
Både B-type Natriuretic Peptide (BNP) og N-terminal pro-BNP (NT-proBNP) kan brukes i Mayo prognosemodell, men en må oppmerksom hvilken som benyttes og korrekt referanseverdi. Det samme gjelder bruk av Troponin T og Troponin I i revised Mayo prognosemodell.
Behandling og prognose påvirkes av andelen av plasmaceller i benmarg (Kourelis et al., 2013; Pardanani et al., 2003). Pasienter med mer enn 10 % plasmaceller uten at kriteriene for behandlingskrevende myelomatose er oppfylt, har dårligere prognose. Funn av t (11;14) er assosiert med redusert PFS og dårligere klonal respons på bortezomib og IMIDs og dårligere overlevelse. Del17p er også assosiert med dårlig prognose og dårlig terapi respons ved AL amyloiose.
Tabell 9.2: Mayo-stage (2004), Mayo2004/European og Revised-Mayo prognosemodeller
dFLC er differanse mellom involverte og ikke-involverte immunglobulin lettekjede (eks verdi kappa minus verdi lambda)
Stadium | Kriterier | Median overlevelse |
|---|---|---|
Mayo-stage (2004) | ||
Stadium I | NT-proBNP <332ng/L (BNP 81 ng/L) og TnT <35ng/L (TnI 100 ng/L) | 26,4 mnd |
Stadium II | NT-proBNP ≥332ng/L eller TnT ≥35ng/L | 10,5 mnd |
Stadium III | NT-proBNP ≥332 ng/L og TnT ≥35ng/L | 3,5 mnd |
Mayo 2004/European: Mayo 2004 stadium III er delt i 2 grupper | ||
Stadium IIIA | NT-proBNP< 8500 ng/L (BNP 700 ng/L) | 15 mnd |
Stadium IIIB* | NT-proBNP >8500 ng/L (BNP 700 ng/L) | 4 mnd |
* Pasienter med NT-proBNP >8500 ng/L og systolisk blodtrykk <100 mmHg har en særskilt dårlig prognose, noe som må vektlegges ved valg av behandling.
Revised Mayo-prognosemodell (2012) | ||
Stadium I | Ingen av følgende tre kriterier oppfylt NT-proBNP<1800 ng/L TnT <25ng/L (TnI 100 ng/L) dFLC <180 mg/L | NR |
Stadium II | Et positivt kriterium | 68,8 mnd |
Stadium III | To positive kriterier | 16,7 mnd |
Stadium IV | Tre positive kriterier | 6,7 mnd |
NT-ProBNP og BNP øker ved nyresvikt.
Førstegangsbehandling av systemisk AL amyloidose
Sist faglig oppdatert: 28.08.2023
Alle pasienter med symptomatisk systemisk AL amyloidose inkludert de med isolert koagulopati eller nevropati skal behandles. Behandling av amyloidose retter seg mot underliggende plasmacelle/B-celle neoplasi med mål om å oppnå «very good partial remission» (VGPR).
Toleransen for behandling er dårligere ved amyloidose enn ved myelomatose og medikamentene må spesielt ved hjerteamyloidose initialt doseres lavere enn ved myelomatose. Dosene kan økes ved god toleranse.
Toleransen for følgende medikamenter må vektlegges under behandling: (se egne kurdefinisjoner i slutten av kapittelet)
Tabell 9.3
| Medikamenter med dokumentert effekt i førstelinjebehandling | |
Medikament: | Forsiktighetsregler: |
Melfalan | Anbefalt maksimal dose 0,22 mg/kg/dag. Ved nyresvikt reduksjon 25–30 %. |
Bortezomib | Økt risiko for arytmier, plutselig hjertedød og polynevropati (også ved sc. administrasjon). Risiko for redusert respons hos pasienter med t (11;14). Anbefalt redusert dose: se tabell 9.7. |
Dexamethason | I dosering 40 mg/dag økt risiko for arytmier og trombose og bør derfor hos pasienter med stadium II–III primært reduseres til 20 mg. |
Daratumumab | Svært godt tolerert. Ikke behov for dosejustering. Kombinasjon daratumumab-CyBorDex er til vurdering av Nye Metoder Daratumumab (Darzalex) - Indikasjon II (nyemetoder.no) |
Medikamenter aktuelle ved behandling av residiv (i tillegg til ovenstående) | |
Lenalidomid/ Pomalidomid | Økt myelosuppressiv effekt. Revlimid tolereres dårlig. Anbefalt maksimal dose initialt er 15 mg/dag. Grunnet betydelig toksisitet anbefales ikke revlimid ikombinasjon med melfalan. Stigning av NT-proBNP og TnT under behandling med IMIDs, og disse kan ikke brukes for evaluering av organrespons (Dispenzieri et al., 2010). Det er uavklart om dette er en direkte kardiotoksisk effekt av IMIDs. Ved klinisk forverring av hjertesvikt under behandling bør seponering av lenalidomid vurderes. |
Behandlingsmål, behandlingslengde og evaluering av respons
Behandling ved amyloidose har følgende hovedmål:
- Rask reduksjon av monoklonalt protein. Dypest mulig respons skal tilstrebes for å forhindre ytterligere fall i organfunksjon, og på sikt bedring av organfunksjon.
- Behandlingen skal være tilpasset og individualisert med henblikk på grad av organdysfunksjon, se tabell 9.3.
- Optimalisert støtteterapi for å minske behandlingsrelatert morbiditet og mortalitet.
Ved amyloidose vurderes både hematologisk respons og respons i de forskjellige affiserte organene.
Vurdering av hematologisk respons
Definisjoner for hematologisk respons ved amyloidose er oppsummert i tabell 9.5.
Absolutt reduksjon i involverte FLC korrelerer med forbedret overlevelse uavhengig av type terapi. Behandlingsmål er minst VGPR.
Evaluering av hematologisk respons og vurdering av behandlingssvikt skal gjennomføres tidlig i behandlingen. Fravær av minst VGPR 100 dager etter HMAS eller etter 2–3 sykluser med kjemoterapi hos pasienter som ikke er kandidater for HMAS, defineres som behandlingssvikt. Hos HMAS-pasienter med behandlingssvikt anbefales konsolidering med bortezomibholdig regime. Hos pasienter som behandles med kun kjemoterapi, må behandling med alternativt 2. linje regime vurderes.
Behandlingslengde
Plasmcellebyrde er generelt lav ved AL amyloidose, og det er som regel ikke behov for like lang behandling som ved myelomatose.
Ved AL amyloidose med plasmacelleandel <10 % vil det være tilstrekkelig med 4–6 kjemoterapisykluser.
Pasienter med AL amyloidose og plasmaceller >10 % bør ha lengre behandling, opptil 6–12 kjemoterapisykluser.
Pasienter med myelomatose og amyloidose skal så lenge det er god toleranse ha behandling etter generelle retningslinjer for myelomatosebehandling med dosejusteringer initialt som ved AL-amyloidose.
Vurdering av organrespons
Definisjoner for respons i forskjellige organer er angitt i tabell 9.6. Organrespons evalueres med NT-pro-BNP for hjerteaffeksjon, reduksjon i albuminuri ved nyreaffeksjon, og redusert ALP samt redusert størrelse av lever på ultralyd/CT. Ekko cor er ikke påkrevd i oppfølging av organrespons. Ekko cor kan være av verdi til sammenligning før behandling på senere tidspunkt.
Organrespons kommer senere enn hematologisk respons, og kan ta opptil flere måneder/år. Fravær av organrespons ved optimal hematologisk respons indikerer derfor ikke behandlingssvikt
Anbefalt hyppighet av responsevaluering
Det vises til tabell 9.8.
Pasienter som er kandidater for HMAS
Sist faglig oppdatert: 28.08.2023
Med mindre det foreligger kontraindikasjoner er høydose melfalan med autolog stamcellestøtte førstevalget for pasienter som er yngre enn 70 år (Sanchorawala et al., 2015).
Pasienter som vurderes for autolog HCT bør oppfyle følgende kriterier:
- Alder <70 år
- PS 0–2
- NYHA klasse I–II
- Systolisk BT >90 mm Hg
- Troponin T <60 ng/L
- NT-proBNP <5000 ng/l
- eGFR> 40 mL/min (for pasienter som ikke er i dialyse)
- bilirubin <1,5 xULN uten tegn til syntesesvikt
Noen pasienter som tidlig i forløpet ikke ansees å være kandidater for HMAS kan etter induksjonsbehandling oppnå betydelig bedring av organfunksjon og dermed bli kandidat for HMAS.
Induksjonsbehandling
Androma studien (Kastritis et al., 2021) viser høyere responsrate og signifikant bedre hendelsesfri overlevelse etter 4 sykluser med Daratumumab - CyBorDex enn ved behandling med bare CyBorDex. Denne kombinasjonen er derfor ansett som den beste behandlingen. Kombinasjonen Daratumumab - CyBorDex er til vurdering hos Nye metoder, Daratumumab (Darzalex) - Indikasjon II (nyemetoder.no). I påvente av beslutningen i Nye metoder, kan fagdirektør søkes om bruk av medikamentet (uavhengig av om det foreligger isolert AL amyloidose eller AL amyloidose med myelomatose). Se Unntaksordning (nyemetoder.no)
Alternativt kan CyBorDex benyttes i tilfeller der det ikke er mulig å gi tillegg av Daratumumab (Mikhael et al., 2012; Venner et al., 2012).
Noen anbefaler at pasienter med isolert nefrotisk syndrom og mindre enn 10 % plasmaceller i benmarg ikke trenger induksjonsbehandling og at man kan vurdere å gå rett til høydosebehandling uten induksjonsbehandling (Dispenzieri et al., 2013). Data indikerer at induksjonsbehandling kan være fordelaktig ved denne pasientgruppen (Huang et al., 2014; Hwa et al., 2016). Induksjonsbehandling er derfor anbefalt til alle pasienter som er kandidat for HMAS.
Mobilisering av stamceller
Stamcellemobilisering gjennomføres ved bruk av G-CSF alene. Mobilisering med G-CSF og cyklofosfamid gir økt risiko for pulmonale og kardiale komplikasjoner og bør derfor ikke benyttes.
G-CSF doseres 10 µg/kg/døgn fordelt på 2 doser i 4 påfølgende dager.
Melfalan og infusjon av stamceller
Melfalan doseres 200 mg/m2 med GFR >30 ml/min/1,73 m2 (Cibeira et al., 2011). Pasienter med GFR <30 ml/min/1,73 m2 som ikke er i dialyse er ikke kandidat for HMAS. Hos pasienter i dialyse doseres Melfalan 140 mg/m2.
Infusjon av minst 2,5x106 CD34+ celler per kg pasientvekt (helst 4 til 5 x106 CD34+ celler per kg pasientvekt) 24–48 timer etter infusjon av melfalan. Pasienter med hjerteaffeksjon skal observeres med telemetri under og en time etter stamcellereinfusjon.
Grunnet økt risiko for pulmonale komplikasjoner og engraftment syndrom anbefales ikke rutinemessig bruk av G-CSF i aplasiperioden (Comenzo et al., 2002).
Oppfølging etter HMAS
Pasienter som ikke oppnår minst VGPR 100 dager etter HMAS ansees ikke å ha tilstrekkelig respons (Landau et al., 2013). Det anbefales konsoliderende behandling med bortezomib holdig regime.
Pasienter som ikke er kandidater for HMAS
Ca. 75 % av alle pasienter med amyloidose vil på grunn av alder og/eller uttalt organaffeksjon ikke være kandidater for HMAS. Det finnes en rekke regimer som er forsøkt som førstelinje ved amyloidose. Basert på resultater av nylig gjennomført Andromeda studie, anbefales bruk av daratumumab- CyBorDex til disse pasienter. Det er for øvrig svært få andre studier som direkte har sammenlignet forskjellige regimer. Alternativt kan MelBorDex eller CyBorDex benyttes hvor dosering av de forskjellige medikamentene vil avhenge av Mayo stadium og komorbiditet og er oppsummert i tabell 9.7.
Kombinasjon daratumumab-CyBorDex er til vurdering av Nye Metoder.
Pasienter med Mayo stadium I
Pasienter i denne kategorien har ingen eller lite affeksjon av hjertet. Disse pasientene forventes å ha god toleranse for fulldosert trippelbehandling. Fulldosert daratumumab- CyBorDex er førstevalg. MelBorDex eller MelBorDex, er alternative regimer hvis daratumumab ikke er tilgjengelig. Hos pasienter som kan bli aktuelle for HMAS på et senere tidspunkt skal ikke MelBorDex benyttes.
Mayo stadium II/IIIA
Disse pasientene har nytte av trippelbehandling, hvis doser justeres adekvat. Førstevalg hos disse pasientene vil være daratumumab- CyBorDex.
MelBorDex med dosejustering av Melfalan, bortezomib og dexamethason. Alternativ er dosejustert CyBorDex.
Hos pasienter som kan bli aktuelle for HMAS på et senere tidspunkt skal ikke MelBorDex benyttes.
Hos pasienter i stadium I eller II med uttalt polynevropati kan man vurdere cyklofosfamid/lenalidomid/dexamethason, MelDex, PomDex eller annen daratumumab holdig regime.
Mayo stadium IIIB med eller og uten ortostatisk hypotensjon
Disse pasienter har en svært dårlig prognose.
Paseinter med AL amyloidose i stadium IIIB var ikke inkludert i Andromeda studie. Grunnet mulighet for økt kardiell toksisitet og manglende data med tanke på sikkerhet kan kombinasjonen daratumumab- CyBorDex foreløpig ikke anbefales for denne pasientgruppe. Anbefalt førstelinjebehandling er doseredusert MelBorDex. Behandling bør foregå inneliggende med telemetriovervøkning etter de første dosene med bortezomib.
IgM assosiert amyloidose eller amyloidose assosiert med lymfoproliferativ sykdom
Behandlingen rettes mot patogen klon, det vises til kapittel for Mb. Waldenström og KLL.
Vedlikeholdsbehandling
Pasienter med myelomatose og amyloidose skal følge retningslinjer for vedlikeholdsbehandling, for tiden er dette ikke godkjent av Nye Metoder. For pasienter uten myelomatose anbefales ikke vedlikeholdsbehandling.
Tabell 9.4
Risikogruppe | Anbefalt førstelinjebehandling | Alternativ regime |
|---|---|---|
Stadium I/II, NT-proBNP <5000 ng/L | Dara-CyBorDex | MelBorDex CyBorDex 1 |
Stadium II, Ingen polynevropati | Dara-CyBorDex | MelBorDex (doseredusert) CyBorDex 1 |
Ved med uttalt neuropati Uansett stadium | MelDex | CyLenDex; LenDex; PomDex, daratumumab baserte regimer |
Stadium IIIA | Dara-CyBorDex | MelBorDex (doseredusert) CyBorDex (doseredusert) |
Stadium IIIB (proBNP> 8500 ng/L, | MelBorDex (doseredusert) oppstart inneliggende, vurder telemetri-overvåkning | CyBorDex (doseredusert) |
1Bør benyttes hvor man er usikker på om HMAS kan være aktuelt på senere tidspunkt
Adjuvant behandling
Sist faglig oppdatert: 23.12.2021
Kontinuerlig behandling med doksysyklin hemmer fibrillogenese og 100 mg to ganger daglig kombinert med standard kjemoterapi har i en retrospektiv analyse vist overlevelsesgevinst hos pasienter med hjerteamyloidose (Wechalekar et al., 2017). Effekten var signifikant ved stadium III med pro-BNP under 8500ng\L og uten ortostatisk hypotensjon. Imidlertid er kostnad/bivirkningsprofilen for doksysyklin lav og man kan forsøke dette som adjuvant behandling ved hjerteamyloidose.
Behandling av residiv
Sist faglig oppdatert: 28.08.2023
Det et er ingen konsensus internasjonalt om hva som defineres som behandlingskrevende residiv og andrelinjebehandling må veies opp mot komorbiditet, alder og forventet toksistet ved behandling.
Det er generell enighet om at behandling indisert ved hematologisk tilbakefall og før det har tilkommet organskade. Hovedproblemet er at det mangler validerte kriterier for hematologisk tilbakefall.
I fravær av klare retningslinjer anbefales oppstart av residiv behandling i følgende situasjoner:
- ved tegn til hematologisk tilbakefall etter oppnådd optimal respons ved tidligere behandling uavhengig om det er tegn til ny/ forverring av organskade. Hematologisk tilbakefall etter oppnådd optimalt respons (minst VGPR) er definert som:
- tilkommet M-komponent hvis denne ikke var til stede etter tidligere behandling
- abnormal FLC ratio og minimum dobling av involverte FLC
- AL amyloidose relatert organprogresjon uten hematologisk progresjon. Alternative årsaker til organskade bør utelukkes og tilstedeværelse av amyloid klon bør verifiseres.
- Pasienter med dFLC<50 mg/L ved diagnose lar seg ikke vurdere ihht de validerte kriteriene for VGPR (eller PR)- Disse har i utgangspunktet god prognose, men dersom de har hatt alvorlig organaffeksjon ved diagnose med lav lett kjedebelastning ved diagnose, bør 2. linjes behandling vurderes ved kun minimal stigning i dFCL fra NADIR etter 1. linje.
- Pasienter som har hatt alvorlig hjerteaffeksjon definert som Mayo stadium II–III ved diagnose bør vurderes for 2. linjes behandling tidlig og før kriteriene for hematologisk tilbakefall er oppfylt.
Behandling ved residiv
Daratumumab gir rask og dyp respons og er godt tolerert. Daratumumab baserte regimer kan benyttes ved residiv (Kaufman et al., 2017; Sher et al., 2016). Best effekt sees der daratumumab ikke er benyttet i første linje eller om det har gått mer enn 2 år siden forrige behandling med daratumumab. Hos pasienter med residiv som ikke er refraktære for daratumumab er det enighet om at daratumumab ansees som den beste behandlingen. Daratumumab har ikke markedsføringstillatelse for denne indikasjonen og er ikke meldt inn til vurdering i Nye metoder.
Hos pasienter der behandling med daratumumab ikke er tilgjengelig er følgende regimer mulig (Kastritis et al., 2012; Kumar et al., 2012; Palladini et al., 2017; Sanchorawala et al., 2016):
- >2. år etter forrige behandling: vurder gjenta samme regime
- Bortezomib refraktære: LenDex, PomDex, CyLenDex
- Ikke bortezomib refraktære: daratumumab-CyBorDex, CyBorDex, MelBorDex
- Ixazomib dexamethason
Pomalidomid tolereres generelt bedre enn lenalidomid.
Generelle retningslinjer for støttebehandling ved hjerteamyloidose
Støttebehandling spiller fundamental rolle i behandling av amyloidose. Mål med støttebehandling er å forbedre livskvalitet og forbedre symptomer.
Pasienter med AL amyloidose har økt insidens av både tachy-, bradyarytmier og plutselig hjertedød. Hovedårsaken til plutselig dødsfall er bradyarytmier og elektromekanisk dissosiasjon. Man bør derfor hos de fleste pasientene med amyloidose vurdere 24-timers EKG/Holtermonitorering ved diagnosetidspunktet og ved synkope eller andre symptomer på bradyarytmi.
Man har ikke kunne vise at primærprofylaktisk implantasjon av ICD gir overlevelsesgevinst. Implantasjon av ICD følger vanlige retningslinjer (Lin et al., 2013).
Pasientene har betydelig økt insidens av atrieflimmer, intrakardiale tromber og hjerneslag. Risiko for hjerneslag er høyere enn det CHADSVA2SC angir for atrieflimmer uten amyloidose (Feng et al., 2007). Forekomst av hjerneslag er også økt hos pasienter hvor man ikke kan påvise atrieflimmer grunnet redusert forkammerfunksjon (Feng et al., 2009). Antikoagulasjon reduserer risiko for tromboembolisk hendelse (Feng et al., 2009). Så lenge det ikke foreligger blødningstendens skal man følge vanlige retningslinjer for antikoagulasjon.
Noen anbefaler antikoagulasjon også hos pasienter uten påvist atrieflimmer ved kun påvist forstørrede forkamre (Feng et al., 2007; Feng et al., 2009).
Symptomatisk hjerteamyloidose behandles først og fremst med loop-diuretika og kaliumsparende diuretika. ACE hemmere, AT2-blokkere og kalsiumantagonister bør unngås eller brukes med forsiktighet; spesielt hos pasienter med autonom neuropati, nyresvikt og hypertensjon. Beta-blokkere kan brukes ved atrieflimmer, men gir økt risiko for hypotensjon og bradyarytmier. Amiodarone er som regel godt tolerert. Grunnet interaksjon mellom amyloidfibriller og digoxin/digitoxin er toleranse for denne medikamentklassen redusert og bør kun brukes med stor forsiktighet (Falk, 2005; Rubinow et al., 1981).
Hjertetransplantasjon kan vurderes hos unge pasienter uten myelomatose som har alvorlig isolert hjertesykdom og som har oppnådd VGPR eller CR
Tabell 9.5: Oppdaterte kriterier for hematologisk respons eller hematologisk progresjon (Gertz et al., 2005)-PMID 33410355
FLC er nivå av involverte frie lette kjeder, dFLC er differanse mellom involverte og ikke-involverte immunglobulin lettekjede (eks verdi kappa minus verdi lambda).
Hematologisk | Kriterier for respons |
|---|---|
CR | Begge kriterier må være oppfylt:
|
VGPR | dFLC <40 mg/L* |
PR | Reduksjon i dFLC >50 % |
Ikke respons | Alle andre pasienter |
* For pasienter med dFLC 20–50 mg/L ved diagnose, reduksjon i dFLC<10 mg/L assosiert med bedre overlevelse
| Kriterier for progresjon |
|---|---|
Hvis CR | detekterbar M-protein eller unormal FLC ratio med minimum dobling av nivå av involverte FLC |
Hvis PR | >50 % dobling i M- komponent til >5 g/l eller >50 % økning i urin M-komponent til over 200 mg/ døgn eller >50 % økning i FLC til >100 mg/l |
Tabell 9.6: Kriterier for organ respons eller progresjon (Comenzo et al., 2012)
Organ | Kriterier for respons | Kriterier for progresjon |
|---|---|---|
Hjerte | Reduksjon i NT-proBNP >30 % eller >300 ng/ng/L (hvis baseline NT-proBNP >650ng/mL) eller ≥2 punkt reduksjon i NYHA klasse (hvis baseline NYHA III eller IV) | >30 % økning i NT-proBNP eller >300 ng/L økning i NT-proBNP eller ≥33 % økning i Tropin-T eller ≥10 % reduksjon i EF. |
Nyre | ≥30 % reduksjon i proteinuri eller Proteinuri ≥0,5 g/24t og fravær av økende nyresvikt definert som >25 % reduksjon i eGFR | ≥50 % (>g/L per d) økning i 24 t urin-proteinutskillelse eller ≥25 % stigning i kreatinin eller ≥25 % reduksjon i baseline kreatinin |
Lever | ≥50 % reduksjon i ALP verdi eller reduksjon av leverstørrelse ved radiologisk undersøkelse ≥2cm. | ≥50 % stigning i ALP fra laveste registrerte verdi |
Nerver | Bedring bekreftet ved elektromyografi | Progredierende nevropati bekreftet ved elektromyografi |
Tabell 9.7: Oppsummering av de forskjellige behandlingsregimene
Dara-CyBorDex: Daratumumab- Cyklofosfamid, Bortezomib, Dexamethason | ||
Sykluslengde 28 dager | ||
Medikament | Dosering | Dag |
Daratumumab subkutant | 1800 mg | Syklus 1–2: dag 1,8,15,22 Syklus 3–6: dag 1,15 Deretter hver 4. uke til max 24 sykluser (total behandlingstid) |
Bortezomib subkutant | Dose etter stadium Stadium I: 1,3 mg/m2 Stadium II: 1,0-1,3 mg/ m2. Stadium III: 0,7–1, 0 mg/m2 | Syklus 1–6: dag 1, 8, 15 og 22 |
Cyklofosfamid peroralt | 300 mg/m2 maks 500 mg | Syklus 1–6: 1,8,15,22 |
Dexamethason peroralt* | 40 mg | Syklus 1–6: 1, 8,15, 22 Deretter 20 mg som premedikasjon før daratumumab |
* Når gitt sammen med daratumumab: dexamethason 20 mg dagen med daratumumab behandling og Dexametason gis kun på dag 1 syklus 1 og deretter økes til full dose ved god toleranse. Ved Mayo st. III: dexametason gis kun på dag 1 og 8 og deretter økes til full dose ved god toleranse Vurder reduksjon av cyklofosfamid på 25 %. ved eGFR <30 mL/min/1.73 m2. | ||
CyBorDex: Cyklofosfamid-Bortezomib-Dexamethason | ||
Sykluslengde 35 dager | ||
Medikament | Dosering | Dag |
Bortezomib subkutant | Dose etter stadium Stadium I: 1,3 mg/m2 Stadium II: 1,0–1,3 mg/ m2. Stadium III: 0,7-1, 0 mg/ m2 | Dag 1, 8, 15 og 22 |
Cyklofosfamid peroralt | 350 mg/m2 maks 500 mg | 1,8,15,22 |
Dexamethason peroralt | 20 mg | 1,2,8,9,15,16, 22,23 |
Dexametason gis kun på dag 1 syklus 1 og deretter økes til full dose ved god toleranse. Ved Mayo st. III: dexametason gis kun på dag 1 og 8 og deretter økes til full dose ved god toleranse Vurder reduksjon av cyklofosfamid på 25 %. ved eGFR <30 mL/min/1.73 m2. | ||
MelBorDex: Melfalan-Bortezomid-Dexamethason | ||
Sykluslengde 28 dager | ||
Medikament | Dosering | Dag |
Bortezomib intravenøst | Doses etter stadium Stadium I: 1,3 mg/m2 Stadium II: 1,0–1,3 mg/ m2. Økes ved toleranse Stadium III: 0,7–1,0 mg/ m2 Økes ved toleranse | 1,8,15,22 |
Melfalan peroralt | 0,22 mg/kg | 1,2,3 og 4 |
Dexamethason | 20 mg | 1,2,8,9,15,16, 22,23 |
Dexamethason reduseres til 20 mg kun dag 1 og 8 hos pasienter med uttalt hjertesvikt perifere ødemer, gjentatte arytmier eller vektoppgang >3 %. Kan evt. økes til vanlig dose ved god toleranse Melfalan: 25 % dosereduskjon ved eGFR <30 mL/min/1,73 m2. | ||
MelDex: Melfalan-Dexamethason | ||
Sykluslengde 28 dager | ||
Medikament | Dosering | Dag |
Melfalan peroralt | 0,22 mg/kg p.o | 1,2,3 og 4 |
Dexamethason | 20 mg | 1,2,8,9,15,16, 22,23 |
Dexamethason reduseres til 20 mg dag 1 og 8 hos pasienter med uttalt hjertesvikt perifere ødemer, gjentatte arytmier eller vektoppgang >3 %. Kan evt. økes til vanlig dose ved god toleranse Melfalan reduseres med 25 % ved eGFR <30 mL/min/1,73 m2. | ||
CyLenDex: Cyklofosfamid-Lenalidomide-Dexamethason | ||
Sykluslengde 28 dager | ||
Medikament | Dosering | Dag |
Lenalidomide peroralt | 15 mg | Kontinuerlig dag 1-21 |
Cyklofosfamid peroralt | 500 mg | Dag 1.8.15 |
Dexamethason peroralt | 40 mg | Dag 1,8,15,22 |
Dosereduksjon til 20 mg kun dag 1 og 8 vurderes ved betydelig væskereduksjon, betydelig hjerteaffeksjon og hos eldre. Kan økes ved god toleranse Tromboseprofylakse med albyl-e eller lavmolekylært heparin. | ||
MelLenDex: Melfalan-Lenalidomide-Dexamethason: | ||
Gjentas hver 28. dag | ||
Medikament | Dosering | Dag |
Lenalidomide peroralt | 15 mg | Kontinuerlig dag 1-21 |
Melfalan peroralt | 0,15 mg/kg | Dag 1,2,3 og 4 |
Dexamethason peroralt | 40 mg | Dag 1,8,15,22 |
Dexamethason reduseres til 20 mg kun dag 1 og 8 hos pasienter med uttalt hjertesvikt perifere ødemer, gjentatte arytmier eller vektoppgang >3 %. Kan økes ved god toleranse Melfalan reduseres med 25 % ved eGFR <30 mL/min/1,73 m2. Tromboseprofylakse med albyl-e eller lavmolekylært heparin | ||
Len-Dex: Lenalidomid –Dexamethason | ||
Sykluslengde 28 dager | ||
Medikament | Dosering | Dag |
Lenalidomid peroralt | 15 mg | Kontinuerlig dag 1-21 |
Dexamethason peroralt | 40 mg | dag 1,8,15,33. |
Dexamethason reduseres til 20 mg kun dag 1 og 8 hos pasienter med uttalt hjertesvikt perifere ødemer, gjentatte arytmier eller vektoppgang >3 %. Kan økes ved god toleranse Dosering ved nyresvikt: Tromboseprofylakse med albyl-e eller lavmolekylært heparin | ||
Pom-Dex: Pomalidomid –Dexamethason | ||
Sykluslengde 28 dager | ||
Medikament | Dosering | Dag |
Pomalidomid peroralt | 4 | Kontinuerlig dag 1-28 |
Dexamethason peroralt | 40 mg | dag 1,8,15,22. |
Dexamethason reduseres til 20 mg kun dag 1 og 8 hos pasienter med uttalt hjertesvikt perifere ødemer, gjentatte arytmier eller vektoppgang >3 %.Kan økes til vanlig dose ved god toleranse. Tromboseprofylakse med albyl-e eller lavmolekylært heparin | ||
Daratumumab | ||
Medikament | Dosering | Dag |
Daratumumab subkutant | 1800 mg | Ukentlig første 8 uker Neste 8 doser hver 2.uke Neste 8 doser hver 4. uke |
Ikke behov for noe dosejustering ved redusert nyre, lever eller hjertefunksjon. | ||
Mobilisering med G-CSF | ||
Medikament | Dosering | Dag |
Filgrastim subkutant | 10 µg/kg fordelt på 2 doser Pasienter <70 kg 300 µg morgen og kveld; Pasienter >70 kg 480 µg morgen og kveld; | Høsting av stamceller etter 4–5 dager med G-CSF. |
Mobilisering gis alltid uten cyklofosfamid ved ikke samtidig myelomatose. | ||
Melfalan høydose | ||
Medikament | Dosering | Dag |
Melfalan intravenøst | 200 mg/m2 | To dager før infusjon av stamceller |
| ||
Hos pasienter i dialyse | Melfalan 140 mg/m2 |
|
Tabell 9.8 Anbefalt undersøkelser ved responsevaluering under og etter behandling
| Kjemoterapi | ASCT | Oppfølging | ||
Etter hver syklus | Etter siste syklus | Dag +100 | Hver 2. mnd | Hver 6. mnd | |
Hematogram | x | x | x | x |
|
Nyrefunksjon | x | x | x | x |
|
Leverfunksjon | x | x | x | x |
|
S-elektroforese med immunfiksasjon | x | x | x | x |
|
S-FLC | x | x | x | x |
|
U-elfekstroforese med immunfiksajon |
| x | x |
| x |
Urin albumin/kreatinin ratio |
| x | x |
| x |
Klinisk undersøkelse | x | x | x | x | x |
Kronisk myelogen leukemi (KML)
Sist faglig oppdatert: 23.12.2021
Anbefalinger:
- Ved behandlingsoppstart er det viktig å bestemme et mål for behandlingen, blant annet basert på pasientens alder, komorbiditet og evt fertilitetsønske. TKIene imatinib, nilotinib, dasatinib og bosutinib er førstevalgsbehandling for pasienter med KML (evidensgrad A).
- 2TKIene dasatinib, nilotinib og bosutinib er førstevalg ved imatinib-resistens eller –intoleranse (evidensgrad B). 2TKI induserer varige responser hos noe mindre enn halvparten av pasienter med resistens. Ponatinib, et potent middel som virker på mange punktmutasjoner er et alternativ ved svikt på 2TKI. Mulighet for allo-SCT bør utredes og overveies om andrelinjebehandlingen ikke er optimal (evidensgrad C).
- Diskuter alle nydiagnostiserte pasienter med KML med kolleger ved universitetssykehus, slik at et oppdatert kontroll- og behandlingsopplegg kan iverksettes og at pasientene kan få tilbud om deltakelse i studier (evidensgrad C).
- Effekten av TKI med RT-qPCR ved gitte milepæler er prognostisk viktig (se Behandling av MPN, avsnitt "Ruxolitinib").
- Langvarig dyp molekylær respons er en forutsetning for seponeringsforsøk for å oppnå behandlingsfri remisjon.
- Ved diagnose/debut i blastfase vil man oftest oppfatte tilstanden som akutt myelogen eller lymfatisk leukemi og behandle etter handlingsprogram for hhv. AML/ALL. Påvises BCR-ABL eller Ph+, bør man i tillegg samtidig gi høydosert TKI (evidensgrad B). Enkelte pasienter med debut i akselerert fase har optimal respons på TKI, resten bør vurderes for stamcelletransplantasjon opp til 70–75 års alder.
Hovedsakelige endringer fra tidligere handlingsprogram:
- Risikoskår: Kun ELTS skal brukes
- Karyotype bestemmes kun ved debut. Respons vurderes med RT-qPCR
- Behandlingssvikt etter 12 måneder og senere defineres som transkriptnivå >1 %, siden dette nivået har best korrelasjon med overlevelse
- Behandlingsfri remisjon (TFR) er et realistisk behandlingsmål for
mange pasienter - Ponatinibs rolle nyanseres
Bakgrunn
Sist faglig oppdatert: 23.12.2021
Retningslinjene følger i stor grad anbefalinger fra European LeukemiaNet (ELN) 2020 som bygger på tidligere ELN-anbefalinger (Baccarani et al., 2013; Hochhaus et al., 2020). Det diagnostiseres omtrent 50 nye tilfeller av KML i Norge per år. Insidensen er knapt 1:100 000 og er høyest omkring 60 års alder. Flere enn 90 % av pasientene har et abnormt lite kromosom 22, det såkalte Philadelphia-kromosomet (Ph), som oppstår ved en balansert translokasjon mellom kromosom 9 og 22. Resultatet av translokasjonen er et hybridgen, BCR-ABL, som koder for fusjonsproteinet bcr-abl, med høy tyrosinkinaseaktivitet. Dannelsen av onkogenet BCR-ABL er nødvendig og tilstrekkelig for å utvikle KML.
Diagnose
Sist faglig oppdatert: 23.12.2021
Symptomer og kliniske funn
KML er en aktuell diagnose ved leukocytose, trombocytose, splenomegali eller allmennsymptomer. Omtrent halvparten diagnostiseres tilfeldig i forbindelse med blodprøvetaking av andre grunner. Symptomer kan mangle eller være uttalte, med slapphet, feber, nattesvette, blødningstendens, vekttap og eventuelt skjelettsmerter og tyngdefornemmelse under venstre kostalbue. Mange pasienter har palpabel milt og lever. Lymfeknutesvulst er sjelden (Wisløff et al., 2014).
Ubehandlet KML gjennomgår vanligvis tre faser, kronisk fase (gjennomsnitt 3–4 år), akselerert fase (1/2–1 år) og blastfase (kort levetid). Progresjon er forbundet med økende behandlingsresistens og symptomer. Laboratoriemessig øker antall blaster og basofile granulocytter i perifert blod og ofte oppstår behandlingsrelaterte cytopenier. Blastfase er morfologisk og klinisk en akutt leukemi der immunfenotypen kan være lymfoblastisk eller myeloblastisk (lymfoid eller myeloid blastkrise). Median overlevelse er da kort. Enkelte pasienter debuterer i avansert fase.
Diagnostiske prosedyrer
Miltens størrelse vurderes klinisk i antall cm vinkelrett på ribbensbuen til miltpolen. Nesten alle pasientene har leukocytose, oftest 100–300 x109/L. I tidlig fase kan det bare være lett venstreforskyvning i blodutstryket, men vanligvis sees metamyelocytter, myelocytter og enkelte promyelocytter og myeloblaster. Kjerneholdige erytrocytter er vanlig. Basofile granulocytter er nesten alltid økt. Eosinofile granulocytter og monocytter er som regel også økt i antall, mens lymfocyttallet er normalt. Lett dysplasi er ikke uvanlig. Prosent blaster, eosinofile og basofile granulocytter i perifert blod skal registreres fordi det har prognostisk betydning. Ca. 50 % av pasientene har trombocytose, som kan være isolert, og derfor gjør det nødvendig å utelukke KML i utredning av trombocytoser suspekt på MPN. Anemi er vanlig og kan være uttalt. Forandringene i blodutstryket er vanligvis diagnostiske. Beinmargen er hypercellulær og dominert av venstreforskjøvet myelopoese, men blasttall over 5 % er sjeldent i kronisk fase. I biopsimateriale sees det ofte megakaryocytthyperplasi og av og til fibrose. Dry tap er ikke uvanlig.
Beinmarg sendes til cytogenetisk analyse («G-banding», karyotypering) og perifert blod til molekylærgenetisk undersøkelse på hybridgenet BCR-ABL med polymerasekjedereaksjon (PCR).
Kvalitativ BCR-ABL-PCR påviser p 210/major BCR-ABL1 fusjoner (varianter av e13a2 og e14a2) som forekommer i 95 % av KML tilfellene. I sjeldne tilfeller kan det foreligge «kryptiske» BCR-ABL gener (dvs usynlige ved cytogenetisk undersøkelse) som ikke fanges opp av rutine PCR testen. Passer det kliniske bildet godt med KML, men cytogenetisk us og PCR er negative, bør man i tillegg rekvirere BCR-ABL fluorescerende in situ-hybridiserings (FISH) analyse for å finne uvanlige fusjoner. Dersom positiv FISH test, kan man eventuelt identifisere BCR-ABL translokasjonsgenets variant (e1a2, e6a2, e8a2, e13a3 eller e14a3) ved mer målrettet PCR undersøkelse. e1a2 er vanligst ved Ph+ ALL og er sjeldent ved KML.
Kvantitativ BCR-ABL PCR (RT-qPCR) benyttes til «measurable residual disease» (MRD) oppfølging av pasienter ved behandling med tyrosinkinasehemmere (TKI) og etter allo-SCT (Cross et al., 2012; Hochhaus et al., 2009). Mutasjoner i BCR-ABL kan forårsake behandlingsresistens og kan detekteres med sekvensering.
Supplerende utredning i spesielle tilfeller
Beinmargsbiopsi: Kun ved dry tap
Immunfenotyping (beinmarg/blod): Kun ved blastfase (lymfoid vs myeloid).
Minimumsutredning
- Anamnese: B-symptomer? Blødning? Plager fra milt? Skjelettsmerter?
- Klinisk undersøkelse: Splenomegali (målt i cm vinkelrett på arcus)? Ekstramedullær sykdom?
- Blodprøver: Hemoglobin, hvite, trombocytter, manuell diff, RT-qPCR for BCR-ABL analyse
- Beinmargsaspirat: Morfologisk vurdering + cytogenetisk undersøkelse (G-båndsanalyse)
- Risikostratifisere: Beregne ELTS skår https://www.leukemia-net.org/leukemias/cml/elts_score/
- Melding til Kreftregisteret via KREMT
- Studie? Kontakt universitetssykehus
Prognose og risikofaktorer for sykdomsprogresjon
Sist faglig oppdatert: 23.12.2021
Alder, miltstørrelse platetall, prosentandel basofile, eosinofile og myeloblaster i perifert blod har prognostisk betydning og danner grunnlag for forskjellige skåringssystemer for pasientens risiko for sykdomsprogresjon. Sokal og ELTS skår anses å være best egnet ved TKI-behandling og brukes mest. ELTS angir risko for død av KML og skal i første rekke benyttes. Sokal er lagt til side fordi mange pasienter med høy risk allikevel har god behandlingsrespons. Behandlingsresponsen er allikevel den mest prediktive prognostiske faktor; Også pasienter med høy risikoskår og optimal respons har gode langtidsresultater (Hughes et al., 2010). Noen cytogenetiske tilleggsabnormiteter til Ph er forbundet med dårligere prognose (Fabarius et al., 2011). Også i KML er det funnet tilleggsmutasjoner kjent fra MDS og klonal hematopoese (CHIP) (Branford et al., 2019) og slike mutasjoner er til en viss grad assosiert med behandlingsresistens og forekommer ofte ved blastkrise. Myeloid mutasjonspanel undersøkelse er allikevel ikke anbefalt i klinisk praksis.
Definisjoner
WHO | European LeukemiaNet (ELN) |
Kronisk fase (CP, chronic phase) | |
Blaster i benmarg <10 %. Ingen av kriteriene for AP/BC angitt under. | Blaster i benmarg <15 %. Ingen av kriteriene for AP/BC angitt under. |
Akselerert fase (AP, accelerated phase) | |
Blaster i blod eller benmarg 10–19 %. Basofile i blod ≥20 %. Persisterende trombocytopeni (<100) ikke relatert til behandling. Nytillkomne cytogenetiske avvik under behandling. Trombocytose (>1 000) som ikke responderer på behandling. Økede miltstørrelse og stigende leukocytter som ikke responderer på behandling. | Blaster i blod eller benmarg 15–29 %. Basofile i blod ≥20 %. Persisterende trombocytopeni (<100) ikke relatert til behandling. Nytillkomne cytogenetiske avvik under behandling. Blaster pluss promyelocytter i blod eller benmarg >30 %, med blaster <30 %. |
Blastkrise (BP, blast phase) | |
Blaster i blod eller benmarg ≥20 %. Extramedullær blastproliferation (ikke i milten). Store foci eller aggregater av blaster i margen. | Blaster i blod eller benmarg ≥30 %. Extramedullær blastproliferation (ikke i milten). |
Vi velger å benytte ELN-kriteriene fordi disse er anvendt framfor WHO-kriteriene i de fleste publiserte studiene som vurderer behandlingseffekter av TKI.
Hematologisk respons (HR) forutsetter Hb >11 g/dl, leukocytter innenfor referanseområdet med <5 % metamyelocytter og stavkjernede nøytrofile granulocytter, ingen blaster i blod, normalt blodplatetall, ikke palpabel milt.
Cytogenetisk respons (CgR) kan være komplett (0 % Ph positive metafaser, CCgR), partiell
(1–35 % Ph positive metafaser, PCgR) eller minor (36–65 % Ph positive metafaser, mCgR). Begrepet «major» cytogenetisk respons (MCgR) omfatter både komplett og partiell respons. Ved RT-qPCR motsvarer CCgR ca. 1 % og 35 % Ph+ metafaser ca. 10 %. I ELN 2020 retningslinjer erstattes nå karyotypering som responsparameter med RT-qPCR, men veldig stor del av KML-litteraturen er basert på cytogenetisk respons. Tilleggsavik i karyotype er en del av risikovurderingen ved diagnose og ved behandlingssvikt. Karyotypering skal gjøres ved diagnose og ved behandlingssvikt.
Molekylær respons (MR): Måling av RT-qPCR for BCR-ABL-transkriptet må brukes for høysensitiv kvantifisering av målbar restsykdom (MRD). Svaret angis som antall BCR-ABL transkripter dividert med antall transkripter av et kontrollgen (ved OUS lab. er det GUS) uttrykt som prosent. Det understrekes at utgangspunktet (100 %) ikke er den enkelte pasients diagnoseverdi, men gjennomsnittet av en standardpopulasjon og uttrykkes på en internasjonal skala (IS), slik at resultater verden over skal bli sammenlignbare (Cross et al., 2012). Det er en forutsetning for klinisk bruk av analysesvar at laboratoriet deltar i internasjonale kvalitetskontroller med akseptable resultater. Begrepet «major molekylær remisjon» (MMR) benyttes dersom mengden transkript er redusert med 3 log (tilsvarende >1000 ganger reduksjon) tilsvarer <0,10 % og omtales heretter som MR3). Ved MR3 på TKI behandling, er sjansen for progresjon svært lav.
Begrepet komplett molekylær remisjon (CMR) kreves at man ikke kan påvise transkript overhodet. Begrepet er forlatt, fordi sensitiviteten i prøven angir hvor negativ en negativ prøve er, og derigjennom reflekteres responsdypet. Begrepene MR4 ≤0,01 %, MR 4.5≤0,0032 % og MR 5≤0,001 % er mer presise og alle disse responsnivåene aksepteres som dyp molekylær respons (DMR), som er godt korrelert med muligheten for varig TFR.
BCR-ABL mutasjonsanalyse: Resistens mot TKI kan skyldes amplifisering eller punktmutasjoner i BCR-ABL genet. Relevante punkt-mutasjoner fører til utbytte av enkelt-aminosyrer, og slike molekylendringer resulterer i opphevet eller lavere affinitet av bindingen mellom TKI og bcr‑abl.
- Det anbefales at klinikeren bestiller mutasjonsanalyse ved stigning av BCR-ABL-transkript med mere enn 5 ganger tidligere nivå (forutsatt at pasienten har transkriptnivå >0,1 % (evidensgrad D).
Denne analysen kan etterbestilles på telefon og utføres i samme prøvemateriale som ble tatt til RT-qPCR for BCR-ABL. Ved funn av punktmutasjoner som gir TKI-resistens, se Førstelinjebehandling i kronisk fase, avsnitt "2TKI (dasatinib, nilotinib og bosutinib) i 1. linje".
Førstelinjebehandling i kronisk fase
Sist faglig oppdatert: 23.12.2021
Strategi ved debut
- Alle pasienter med nydiagnostisert KML bør diskuteres med universitetssykehus, for å lage en individuell behandlingsplan og gi tilbud om studieinklusjon (evidensgrad D).
- Hvilket mål skal oppnås: Overlevelse med god livskvalitet? behandlingsfri remisjon?
- Behandling med TKI bør startes når diagnosen er sikker, selv om pasienten ikke har symptomer (evidensgrad A). Det er i hovedsak to strategier for TKI-behandling.
1) Imatinib fra start og raskt bytte ved dårlig respons (gjelder flertallet av pasienter). 2) Dasatinib, Nilotinib eller Bosutinib for å oppnå dyp respons raskt. Pasientpreferanse, bivirkningsprofil og komorbiditet er viktig for valg av strategi.
Selv pasienter med kort forventet overlevelse uavhengig av leukemien, behandles med TKI, oftest er imatinib mest aktuelt, evt Hydroxyurea (HU)). Dersom det er indikasjon for å starte behandling før diagnosen er bekreftet (leukostase, kraftig trombocytose), kan man velge å starte med HU. HU kan også gis parallelt med TKI initialt om det er behov for å redusere celletallet raskt (dosering se under). Ved start av leukemibehandling oppfordres pasienten til rikelig væskeinntak som tumorlyse-profylakse.
Målet med behandlingen er først og fremst å hindre progresjon av sykdommen og å opprettholde god livskvalitet med langtids overlevelse. Seponering av TKI med nøye oppfølging hos pasienter med spesifisert god respons er nå del av standardoppfølgingen av KML (Mahon et al., 2010). Gir man 2TKI ved diagnose, vil flere oppnå dypere responser raskt og det er rimelig å regne med at denne strategien vil øke andelen pasienter som på sikt kan avslutte behandlingen uten tilbakefall. Denne strategien er mest interessant for yngre pasienter, særlig unge kvinner med evt barneønske. Uavhengig av TKI-valg ved diagnose, vil allikevel de fleste pasienter måtte behandles livslangt med TKI.
Imatinib
Fusjonsproteinet bcr-abl er en konstitutivt aktiv tyrosinkinase som fosforylerer en rekke substrater involvert i KML-cellers vekst, differensiering og apoptose. Hemming av bcr-abl griper i motsetning til cytostatika og interferon (IFN), direkte inn i sykdommens årsak. Den best utprøvde hemmeren er imatinib. Den har revolusjonert behandlingen av KML, tolereres godt og er godt dokumentert førstelinje-behandling (evidensgrad A) (Hochhaus et al., 2009). Ikke alle pasienter har optimal behandlingsrespons, en tredjedel vil trenge annen behandling i et 5-års perspektiv, grunnet resistens eller intoleranse/ bivirkninger (de Lavallade et al., 2008; Hochhaus et al., 2009). Imatinib er registrert i Norge til behandling av voksne pasienter med Ph+ og / eller BCR-ABL-positiv KML i alle sykdomsfaser og er generisk legemiddel. Anbefalt dose er 400 mg/dag i kronisk fase. Høyere initialdose enn 400 mg gir raskere tidlige responser i kronisk fase, men andelen gode respondere blir like i gruppene med tiden.
2TKI (dasatinib, nilotinib og bosutinib) i 1. linje
Effekt og bivirkninger av både dasatinib nilotinib og bosutinib er sammenlignet med imatinib ved nydiagnostisert KML i kronisk fase (Cortes et al., 2018a; Kantarjian et al., 2012a). Alle midler gir raskere og dypere responser enn imatinib, molekylært og cytogenetisk. 2TKI øker andel pasienter med udetekterbar sykdom. Dette kan være et poeng hos pasienter med høyrisiko sykdom, eller yngre pasienter der behandlingsfri remisjon (TFR) er et viktig mål. Spesielt unge kvinner med fertilitetsønske er gode kandidater for å få 2TKI fremfor imatinib for å ha størst mulig sjanse til å gjennomføre graviditet i TFR. Det er ingen sikker overlevelsesforskjell sammenliknet med imatinib, men litt færre progresjoner til avansert fase (ca. 1–2 % mot ca. 4 % første år). Alle midler er godkjent av Statens legemiddelverk til bruk i første linje. Alle medikamentene har noen mulig alvorligere bivirkninger (se avsnitt under). Man står altså relativt fritt (evidensgrad A) i valg av behandling i 1. linje med dasatinib, imatinib, nilotinib og bosutinib, men imatinib er fortsatt meget effektivt, sikkert og velkjent for hematologene
Om pris for legemidler: TKI forskrives på H-resept. Imatinib er generisk og er det kostnadseffektive alternativet. I siste anbudsrunde var «Imatinib Accord» rimeligst og foretrukket. Forskrivere skal være oppmerksom på at det ved H-resept ikke finnes noen ordning for utlevering av billigste alternativ, slik at man må skrive resepten på det eksakte preparatet man mener at pasienten skal ha. Prisene på imatinib-generika varierer betydelig. Apotekene tjener bedre på å utlevere dyrere varianter. Bare spesielle grunner må foreligge for å velge annet enn rimeligste alternativ. Sannsynligvis endres vilkårene ved neste anbud og foreløpig må den enkelte lege følge med på prislistene og endre til billigste variant. Som eksempel kan nevnes at en produsent har vunnet anbudet det ene året for ved neste å øke prisen 10–20 ganger og håper nok på at legene skal glemme å bytte. Foreløpig er nilotinib, bosutinib og dasatinib betydelig dyrere med priser over 400.000/år. Dasatinib er også generisk men har foreløpig med samme pris som originalpreparatet.
Nilotinib (Tasigna®, Novartis): Nilotinib er registrert på indikasjonen KML i alle faser med imatinib intoleranse eller resistens, og også godkjent som førstelinjebehandling av KML i kronisk fase i dosen 300 mg x 2 (Cortes et al., 2018a).
Dasatinib. (Sprycel®, Bristol-Myers Squibb): Dasatinib er registrert på indikasjonen KML i alle faser Dasatinib er ca. 300 ganger mer potent enn imatinib og er effektivt in vitro ved langt de fleste mutasjoner assosiert med imatinibresistens unntatt T315I, F317L og V299L. Dasatinib hemmer i tillegg til abl også flere kinaser i src-familien, samt TEC-kinaser, c-kit og PDGFR. Dasatinib doseres i kronisk fase 100 mg x 1, i akselerert fase og blastfase 140 mg x 1. Erfaringsmessig finnes noen pasienter som har vanskelig for å tåle 100 mg daglig over lang tid, men som kan ha meget god effekt av lavere dose, f.eks 50–70 mg x1.
Bosutinib. (Bosulif, Pfizer): Godkjent i første linje av EMA. Litt raskere respons enn imatinib. Virksom ved mange imatinib-mutasjoner. Mindre tendens til pleuraeffusjon enn dasatinib til tross for at Bosutinib hemmer to av Src-kinasene. Doseres 400 mg daglig i kronisk fase og 500 mgx1 i avansert fase. I likhet med dasatinib finnes mange pasienter med god effekt på lavere dosenivå. Høye transaminaser kan forekomme hos en betydelig andel av pasientene
Respons og monitorering
- Effekten av TKI ved gitte tidspunkter (milepæler) er prognostisk viktig.
Følg hematologiske tellinger og biokjemi for overvåkning av organfunksjoner til stabil hematologisk respons. Erfaringsmessig er det vanligst med myelosuppresjon i 2. eller 3. behandlings måned. Støtte-behandling med G-CSF, evt TPO og/eller transfusjoner bør foretrekkes fremfor dosereduksjon. - RT-qPCR måles i perifert blod hver 3. måned til bekreftet (stabil) MR3, deretter hver 6. måned. Dersom pasienten ikke har oppnådd 10 % BCR-ABL på 3 månedserskontrollen skal man gjenta prøven raskest mulig. Se tabell 10.1 for skille på «advarsel» eller «svikt».
- Kinetikken i responsen etter 3, 6, 12 måneder og deretter, danner grunnlag for å kategorisere pasientens behandlingsrespons som «optimal», «advarsel» eller «svikt». Kravene til responskinetikken gjelder uavhengig av hvilken TKI som benyttes i første linje.
- MR2 (<1 %) oppfattes best korrelert til overlevelse, og MR3 (<0,1 %) oppfattes som trygg havn med leveutsikter som gjennomsnittsbefolkningen.
Tabell 10.1 Monitorering av førstelinjebehandling
Adaptert fra ELNs retningslinjer 2020. Tabellen gjelder første og andre linjes behandling i kronisk og akselerert fase med alle TKI godkjent for førstelinjebehandling.
Tid | Optimal | Advarsel | Svikt |
|---|---|---|---|
Diagnose |
| Høyrisiko cytogenetiske tilleggsavvik + ELTS HR |
|
3 mnd | MR ≤10 % | MR>10 % (gjentas raskt) | MR >10 % (bekreftet) |
6 mnd | MR: ≤1 % | MR: >1–10 % | MR >10 % |
12 mnd | MR <0.1 % | MR >0.1–1 % | MR >1 % |
Ved alle senere tidspunkter | MR <0.1 %* | Tap av respons dvs MR >0,1 % | MR >1 %, Cytogenetiske tilleggsavvik i Resistensmutasjoner |
- MR = molekylær respons målt med RT-qPCR
- Høyrisiko kromosomavvik: Trisomi 8; isokromosom 17 dvs. i(17)(q10); trisomi 19; og ulike varianter av øket antall BCR-ABL-kopier som: Ph duplikasjon, +der(22)t(9;22)(q34;q11) og ider(22)(q10)t(9;22)(q34;q11) Se ref (Fabarius et al., 2011). Translokasjoner som involverer tre kromosomer og gir opphav til BCR-ABL eller tap av et Y-kromosom har ikke prognostisk betydning ved TKI behandling.
- Ethvert tap av tidligere respons er et varsel om resistens som må overvåkes nøye inkludert vurdering a mutasjonsanalyse (se under). Som regel bør responskategorien baseres på mere enn ett analyseresultat før det gjøres endringer i behandlingen.
- Som erstatning for karyotypering som responsevaluering (se forrige handlingsprogram) innføres repetert måling av RT-qPCR så snart som mulig dersom man på måned 3 ikke har 10 % eller bedre. Er det >10 % på repetert måling byttes til annen TKI (definert svikt) Karyotypering kan i denne situasjonen gi tilleggsinformasjon med tilleggsavvik av prognostisk betydning selv om slikt forekommer ganske sjeldent.
* Optimal respons for pasienter som vil oppnå TFR er alltid MR4 eller bedre
«Svikt» betyr at valgt TKI bør byttes til annen TKI samtidig som man gjør mutasjonsanalyse, karyotypering og overveier mulighet for allo-SCT med vevstyping av potensielle donores ev søknad til den nasjonale transplantasjonsgruppen.
«Advarsel» betyr at pasienten sannsynligvis har nytte av å bruke den valgte TKI, men kan ha dårligere prognose enn en pasient med «optimal respons». Følges nøye med tanke på tegn til «svikt» og responstap. Oppfølgingen bør skje med kortere intervaller
«Optimal» betyr at det ikke er fordeler med endring av behandlingen
Compliance: Det er nå godt dokumentert at heller ikke KML-pasienter alltid tar foreskrevet medisindose. Redusert compliance angis som en av de hyppigste årsakene til at responsene på imatinibbehandling ikke er optimal (Marin et al., 2010). Ofte er det lavgradige bivirkninger som gjør at pasienter tar behandlingspauser. Overvei skifte av TKI for å bedre compliance. Nilotinib må tas på fastende mage, hvilket kan oppleves som besværlig av enkelte pasienter. Imatinib og Bosutinib tas med stort måltid.
Resistens: Manglende hematologisk eller molekylær respons etter oppstart av TKI er meget sjeldent, mens for treg cytogenetisk respons er mer vanlig. Vanligere opptrer resistens ved at tidligere ervervet respons (hematologisk, CgR eller MR) går tapt unntatt ved compliance-problemer.
Årsakene til TKI-resistens er flere (Apperley, 2007). Punktmutasjoner i BCR-ABLs kinasedomene, amplifisering av BCR-ABL genet, økt transkripsjonsaktivitet, farmakokinetiske faktorer, redusert influx av imatinib (polymorfismer i organisk kation transporter (OCT-1), økt efflux gjennom multi drug resistenspumpen MDR1 / P-glycoprotein og klonal evolusjon i Ph+ klon er assosiert med resistens. De andre TKIene er mindre avhengige av membranpumper og færre ABL-mutasjoner gir resistens. Andre resistensmekanismer enn punktmutasjoner er ikke godt kartlagt Punktmutasjoner utgjør den viktigste resistensmekanismen vi kan påvirke med behandling.
Håndtering og vurdering av kategoriene «Svikt» og «Advarsel»
Sist faglig oppdatert: 23.12.2021
- Compliance. Diskuter bivirkninger og medikamentrutiner med pasienten.
- ABL-mutasjonsanalyse/ karyotypering.
- Farmakokinetiske faktorer / interaksjoner.
- Tettere monitorering.
- Skifte TKI (se avsnittet Valg av 2TKI)
- Evt doseøkning
Primær- eller sekundær «svikt»: Indikasjon for å skifte behandling
- Behandling skiftes til hvilken som helst 2TKI dersom første valg var imatinib eller en annen 2TKI dersom førstevalg var dasatinib eller nilotinib. (evidensgrad B). Ta hensyn til evt mutasjoner og evt komorbiditet (Soverini et al., 2011) (se kapittel Førstelinjebehandling i kronisk fase, avsnitt "2TKI (dasatinib, nilotinib og bosutinib) i 1. linje". Når man har prøvd to 2TKIer er sjansene for meningsfull respons relativt små, kanskje 15-20 % (Hochhaus et al., 2020). Ponatinib gir noe høyere responser, men denne fordelen må veies mot risiko for atheroskleroseassosierte hendelser som potensielt kan gi permanent organskade. I kronisk sykdomsfase er det i retrospektive analyser bedre overlevelse med Ponatinib enn allo-SCT og individuelle avveiinger bør vurderes i samråd med transplantasjonseksperter. Ved resistens i 3. til 4. behandlingslinje er BCR-ABL <1 % et akseptabelt responsnivå.
- Allo-SCT (evidensgrad B) er høyaktuelt dersom respons på 2. linjes behandling eller ponatinib er utilfredsstillende
- Utprøvende behandling i studie.
- Palliativ behandling
Håndtering av punktmutasjoner som gir TKI-resistens
Punktmutasjoner i genet som koder for den ATP-bindende lommen i abl kan føre til TKI-resistens. Det er nå over 100 slike ABL-mutasjoner beskrevet (Soverini et al., 2011). De klinisk relevante mutasjonene fører til bytting av aminosyrer som enten sterisk hindrer TKI-binding (f.eks T315I) eller påvirker bcr-abl konformasjon slik at TKI ikke kan binde. Dasatinib, nilotinib og bosutinib har mindre krav til konformasjonen av bcr-abl enn imatinib. Derfor er det færre mutasjoner som gir resistens mot 2TKIene. Ponatinib er utviklet for å hemme T315I, som alle andre midler er ineffektive mot. Ponatinib virker godt på T315I men også en rekke andre mutasjoner eller andre resistensmekanismer. ABL-mutasjoner er ansvarlige for imatinibresistens i ca. 40 % av tilfellene av behandlingssvikt. De er uvanlige i tidlig kronisk fase, men forekomsten øker med utvikling av sykdommen, sannsynligvis som en manifestasjon av genetisk instabilitet. In vitro data for de forskjellige TKIers effekt ved gitte mutasjoner korrelerer brukbart med klinisk effekt og kan veilede i valg av neste TKI (Soverini et al., 2011). Se tabell i ELNs behandlingsanbefaling fra 2013 (Baccarani et al., 2013). Funn av p-loop ABL mutasjon (aminosyre 248–255) under imatinibbehandling er assosiert med utvikling av blastfase om man ikke skifter behandling. Generelt anbefales det å prøve dasatinib om det foreligger andre mutasjoner enn T315I, F317L eller V299L. Nilotinib vil oftest ikke virke om det foreligger bytting av aminosyre i posisjon 248–255, 315 eller 359. Ved mutasjon bør man alltid vurdereog evt utrede mulighetene for allo-SCT. Påvises mutasjon T315I skal man benytte ponatinib med hensyn til god aterotrombo-profylakse (Cortes et al., 2018b). Alternativer ellers er asciminib og pegylert interferon.
Takling av intoleranse for TKI
Hematologisk toksistet (gjelder alle TKI)
Hematologisk toksisitet er hyppig ved TKI--behandling, antagelig forårsaket av hemming av den bcr-abl-drevne hematopoiesen, og at det følgelig tar tid å gjenreise Ph-negativ, frisk hematopoese. Toleransegrensene for granulo- og trombocytopeni må ses i forhold til totalrisiko og varighet av cytopeni. Opprettholdelse av doseintensitet uten doseopphold er viktig for et godt resultat. Under forutsetning av tett oppfølging i denne fasen, mener vi at doseintensiteten bør opprettholdes så lenge granulocyttallet er over 0.5 x109/L og trombocyttallet ikke faller under 30 x109/L. Kinetikken i fallet av blodlegemer er erfaringsmessig viktig, slik at raske fall ofte vil gi dypere nadir enn langsomme fall. G-CSF / transfusjoner/ TPO-agonister aktuelt (evidensgrad D). Dersom støttebehandling er nødvendig over lang tid, er det grunn til å reevaluere behandlingsopplegget ved å vurdere dosereduksjon eller bytte til annen TKI ved alvorlige eller mindre alvorlige men langtrukne bivirkninger. Nilotinib har mindre hematologisk toksistet enn imatinib, bosutinib og dasatinib noe mer. Anbefalt minimal dose imatinib er 300 mg daglig. Minimal dose er mer usikkert for 2TKIer der doser ned til 200–400 mg x1 (nilotinib) 200 mg x1 (bosutinib) og 20–40 mg x1 (dasatinib) har fått pasienter gjennom cytopenifasen. Så lenge man monitorerer er det et visst spillerom.
Ikke-hematologisk toksistet
Imatinib: Tablettene tas til maten (gjerne største måltid) for å unngå kvalme. Det er relativt få alvorlige bivirkninger. Fatal levernekrose er rapportert og gjør at hepatotoksisk medikasjon inkludert paracetamol, må brukes med forsiktighet. Imidlertid er et stort antall bivirkninger som kvalme, muskelkramper, hodepine, myalgi, artralgi, og utslett beskrevet. Enkelte tilfeller av alvorlig ødemtendens (lungeødem, pleuravæske, ascites, betydelig vektoppgang) har vært observert. Aktiv symptomatisk behandling av smerter (analgetika), perifere ødemer (diuretika, væskerestriksjon), kramper (tøyninger), utslett (lokale eller systemiske steroider), diaré (antidiarrhotika) og kvalme (tas med mat, Afipran) er aktuelt (evidensgrad D). For praktisk håndtering av imatinibbivirkninger og interaksjoner, se Felleskatalogen og (Steegmann et al., 2016).
Nilotinib: Pasientene tåler ofte nilotinib subjektivt bedre enn imatinib. Leveraffeksjon, biokjemisk pankreatitt og stigning av bilirubin hos pasienter med Gilbert genotype er ikke uvanlig (Cortes et al., 2018a). Hudbivirkninger er vanlige. Forverring av diabetes og hyperkolesterolemi er rapportert. Akselerert aterosklerose (underekstremiteter, slag, angina og hjerteinfarkt) er beskrevet hos en mindre andel nilotinibbehandlede pasienter og gir grunn til bekymring i forhold til langtids behandling. Pasienter med diabetes, hypertonikere, røykere og med manifest atherosklerose er sannsynligvis mer predisponert. Alder er sannsynligvis viktig. God behandling av diabetes, hypertoni, atherosklerosemanifestasjoner og røykestopp er logisk å tilby. Veiing av nytte mot risiko i forhold til pasientens kliniske situasjon må gjøres. Nilotinibdosen bør være 300 mg x 2, men ved dårlig respons kan dosen økes til 400 mg x2. Denne høyeste dosen gir en tydelig økning av risiko for slike ikke-reversible bivirkninger.
Dasatinib: Pasientene tåler ofte dasatinib subjektivt bedre enn imatinib. Dasatinib kan et pleuropulmonalt syndrom med immunaktivering som kan gi pleuravæske, perikardvæske, lungeinfiltrater og dyspné hos så mange som 20 % av pasientene i et treårs perspektiv (Kantarjian et al., 2012a). Høyere alder, hjerte-lungesykdom og pasienter med autoimmune sykdommer synes å være predisponert. Pleuratapping kan bli nødvendig, men syndromet er ofte følsomt for behandlingspause og steroider. Tegn på væskeretensjon og lungesymptomer bør tas alvorlig og raskt utredes med røntgen eller CT thorax. Dasatinib-assosiert lymfocyttær serositt (oftest pleuritt) er assosiert med god antileukemisk respons. Man kan ofte tåle noen ukers pause i behandlingen uten vesentlig responstap. Det er også beskrevet tilfeller av reversibel økning av lungearterietrykket (pulmonal arteriell hypertensjon, PAH). Dersom pasienter har dyspnoe, bør man utelukke pleuritt og utrede mht PAH med ekkokardiografi eller invasiv trykkmåling. Endelig er kolitt og GI-blødning hyppigere enn ved andre TKIer.
Bosutinib: Bosutinib gir ofte gastrointestinale ubehag med kvalme og diaré, evt brekninger (Cortes et al., 2012). Disse er ofte overgående og kortvarige, men kan være kraftige. Med forsiktig dosering og symptomatisk behandling kan de fleste pasienter fortsette. Leveraffeksjon er vanlig med bosutinib og må følges opp med blodprøver. Dose 400 mg daglig med mat. Mange velger nå å gjøre en gradvis opptrapping fra 200 mg x1 til 400 mg x1 etter toleranse i løpet av noen uker. Mange pasienter har utmerket antileukemisk respons på 300 mg daglig. Gi høyeste tolerable dose
Ponatinib: Ponatinib gir i likhet med nilotinib, men mer uttalt, atherosklerotiske hendelser som angina, hjerteinfarkt, TIA, hjerneslag og perifere arterielle okklusjoner som kan gi sekvele. Stigning av lever- og pancreas-enzymer er heller ikke uvanlig (Cortes et al., 2018b). Mange pasienter utvikler hypertoni (ofte behov for flere blodtrykksmidler), diabetes og hyperlipidemi. Dette bør helt klart monitoreres og behandles proaktivt (evidensgrad D). Nylig ble foreløpige data av OPTIC-studien forevist på EHA-møtet 2020. Her ble pasientene satt på ulike start-doser. Konklusjonen er at man bør starte med 45 mg daglig og dersom god respons (MR<1 %) reduseres til 15 mg under fortsatt nøye observasjon (Cortes et al., 2020). Ponatinib er det mest potente KML-medikamentet vi har og er det eneste virksomme og godkjente middelet ved mutasjon T315I.
Valg av TKI i annen linje
Det foreligger ikke direkte sammenlignende studier mellom 2TKIer ved imatinibresistens eller intoleranse. Basert på 2 års resultater fra fase II studier (Cortes et al., 2018a; Cortes et al., 2012; Cortes et al., 2018b) synes effekten av medikamentene nærmest ekvivalente og det er rimelig å regne med at man ved imatinibresistens eller intoleranse vil kunne reindusere en varig CCyR hos ca. 40–50 % av pasientene i denne situasjonen.
Dersom pasienten har vært resistent mot 2TKI, er det lav sjanse (ca. 20 %) for akseptabel respons ved å bytte til annen 2TKI. Ved intoleranse er sjansen for repons adskillig bedre. Valg basert på forekomst av resistensmutasjoner bivirkningsprofil, komorbiditet og compliance. Familiedonorsituasjonen bør kartlegges allerede ved behov for annenlinjebehandling hos pasienter med resistens mot 2TKI.
Fertilitet og amming
Sist faglig oppdatert: 23.12.2021
- TKIer er teratogene, embryotoksiske og gjenfinnes i brystmelk. Medikamentene er kontraindisert under graviditet og ved amming (evidensgrad D).
- Interferonbehandling er et alternativ ved graviditet (evidensgrad D).
Enkelte pasienter har gjennomført graviditet etter å ha seponert TKI. Velinformerte pasienter med god respons på TKI (helst minst MR4 i 2 år) kan være kandidater. Dette bør nøye diskuteres med erfarne kolleger. Det er ikke rapportert noen sikker overhyppighet av misdannelser der far er imatinibbehandlet (evidensgrad C) (Abruzzese et al., 2020). Organogenesen foregår i 1. trimester og denne perioden antas å være mest kritisk for misdannelser. TKI-behandling i 3. trimester kan diskuteres dersom rask økning av leukemien. Se referansen for en fin gjennomgang av problemstillinger, også om KML-diagnosen stilles i graviditeten.
Allogen stamcelletransplantasjon
Sist faglig oppdatert: 23.12.2021
Allo-SCT er det eneste dokumenterte kurative behandlingsalternativ for KML, men er forbundet med prosedyrerelatert morbiditet og mortalitet samt en ikke ubetydelig risiko for sene tilbakefall. Det er så langt ikke sikre holdepunkter for at forutgående imatinibbehandling påvirker resultatene av allo-SCT (evidensgrad C). Forutsatt vedvarende kronisk fase etter svikt på imatinib og lav EBMT-score (Gratwohl et al., 2009) (se kapittel Akutt myelogen leukemi (AML)) er resultatene gode (Saussele et al., 2010). De gode resultatene med imatinib har fortrengt allo-SCT som førstelinjebehandling ved KML i kronisk fase.
- Allo-SCT bør alltid overveies som ved svikt på 2TKI (evidensgrad C).
Behandling av akselerert fase
Sist faglig oppdatert: 23.12.2021
- Ved diagnose/ debut i akselerert fase gis 2TKI (evidensgrad C).
- Rekvirer ABL-mutasjonsanalyse primært.
- Pasienter under 70–75 år med stamcelledonor kan være aktuelle for allo-SCT helst etter induksjon av remisjon / kronisk fase (evidensgrad C).
- Dersom pasienten iflg tabell 10.1 responderer «optimalt», kan allo-SCT avventes under forutsetning av god monitorering (evidensgrad C).
Behandling av blastfase
Sist faglig oppdatert: 23.12.2021
- Ved diagnose/debut i blastfase vil man oftest oppfatte tilstanden som akutt myelogen eller lymfatisk leukemi og behandle etter handlingsprogram for hhv. AML/ALL. Påvises BCR-ABL eller Ph+, bør man i tillegg samtidig gi høydose imatinib, dvs 600–800 mg daglig eller 2TKI (evidensgrad C). Dasatinib penetrerer blod-hjernebarrieren, noe som kan være et fortrinn i blastfase. Nilotinib er ikke formelt godkjent for behandling i blastfase, men har omtrent like god effekt som dasatinib. Data på ponatinib i kombinasjon med hyper-CVAD er lovende.
- Ved utvikling av blastkrise under imatinib-behandling gis Ponatinib (TKI behandlingen kan justeres avhengig av ABL mutasjonsstatus og risiko for CNS affeksjon) evt kombinert med konvensjonell induksjonsbehandling for akutt leukemi basert på mutasjonsstatus og immunfenotype (evidensgrad C).
Det er viktig å skille mellom myeloid og lymfatisk blastfase, ved vanlig mikroskopi supplert med immunfenotyping. Myeloid blastfase er hyppigere enn lymfoid (ca. 70 % vs 30 %). All annen behandling enn transplantasjon har kort tidshorisont, men ved lymfoid blastkrise kan man ved oppnådd remisjon kombinere TKI (lovende data med ponatinib, blinatumumab og hyper-CVAD, men også annen mild vedlikeholdsbehandling som steroider, vinkristin, metotrexat, merkaptopurin eller asparaginase (f.eks POMP eller OPAL som er beskrevet i ALL-programmet i palliativ/ transplantasjonsforberedende hensikt) (Jabbour et al., 2015; Sasaki et al., 2016). Allo-SCT forutsetter at ny kronisk fase kan oppnås, donor finnes og at det ikke foreligger absolutte kontraindikasjoner. Transplantasjon i blastkrise inngår ikke i transplantasjonsprogrammet fordi resultatene er meget dårlige. Også med bruk av TKI er resultatene dårlige med svært høye tilbakefallsrater.
Andre behandlingsregimer
Sist faglig oppdatert: 23.12.2021
Hydroksyurea (HU) erstattet busulfan som det viktigste cytostatikum ved kronisk myelogen leukemi, etter at det ble dokumentert lengre overlevelse ved behandling med HU. I tilfeller ved behov for rask reduksjon av antallet leukocytter ved nydiagnostisert KML, kan HU gis sammen med imatinib før senere overgang til imatinib. Øvrig indikasjon for HU vil være behandling hos gamle og svekkede personer samt som palliativ behandling ved svikt på eller intoleranse for TKI dersom pasienten ikke kan transplanteres. Behandlingsmålet er å bringe pasienten i stabil kronisk fase. Standard startdose er 30–40 mg/kg/døgn justert opp eller ned til nærmeste 500 mg (2–4 g/døgn). Når leukocyttene er <20 x109/l, reduseres HU-dosen til forslagsvis 15–20 mg/kg/dag. Videre dosering er individuell. HU har smal terapeutisk bredde, slik at doseforandringer i klinisk rolig situasjon bør skje i relativt små trinn (for eksempel 10–15 % av totaldosen) Gjennomsnittlig vedlikeholdsdose ligger ofte på 1,0–1,5 g daglig der leukocyttall skal holdes mellom 2–5 x 109/l. Laboratoriekontroll anbefales 1–2 ganger per ukentlig initialt, senere sjeldnere. Etter doseendring bør blodverdiene kontrolleres etter 1–2 uker. De fleste pasientene har lite bivirkninger av HU. Kvalme, brekninger og diaré er imidlertid vanlig ved doser over 2 g/dag. Allergiske utslett, aftøse munnsår og hudulcerasjoner inkl leggsår forekommer. Makrocytose og megaloblastisk marg er meget vanlig.
Busulfan (Myleran) er fortsatt aktuelt til noen få pasienter der annen terapi er uegnet. I akselerert fase, der pasienten ikke lenger har effekt av HU, kan busulfan forsøkes. Busulfan brukes også i kondisjonering for allo-SCT.
Interferon: IFN kan prøves ved svikt på TKI, men sjansen for god respons (f.eks. CCgR) er meget lav. Nyere studier gir støtte for at pegylert interferon kan bli et godt tilleggsmedikament til imatinib hos pasienter med KML i kronisk fase men anses fortsatt som eksperimentelt. IFN er et aktuelt medikament om man må behandle KML i graviditet.
Asciminib er en TKI med en svært interessant virkningsmekanisme (Hughes et al., 2019). Vi antar at middelet snart blir godkjent. Asciminib blokkerer ikke det ATP-bindende setet som de andre TKIene, men binder den såkalte myristoyl lommen. Når asciminib binder settes BCR-ABL-molekylet i inaktiv konformasjon. Asciminib virker ved de fleste kjente TKI-resistensmutasjonene inkludert T315I og har god tolerabilitet. På sikt kan man tenke seg at asciminib vil bli brukt på lignende måte som ponatinib i dag. Det vil også komme en studie i første linje der Asciminib prøves mot legens valg av TKI. Middelet kan skaffes på compassionate use.
Seponering-Behandlingsfri remisjon (TFR)
Sist faglig oppdatert: 23.12.2021
Det er nå gjennomført ca. 10 studier av seponering av TKI ved dyp respons, og alle viser langtids TFR på ca. 40-50 % (Laneuville, 2018; Mahon et al., 2010). STIM-studien etablerte prinsippet, og i ettertid er ulike inklusjonskriterier brukt. I den største studien EuroSKI, der 27 norske pasienter deltok, inkluderte man pasienter med MR4 med eller uten detekterbar MRD, mens i mange andre studier var det krav om udetekterbar sykdom og minst MR4.5. MR4, med eller uten påvisbar MRD, ser ut til å være tilstrekkelig dypt (Saussele et al., 2018). Fra EuroSKI og «According to STIM» (aSTIM) har man lært at relaps definert som tap av MR3/MMR er trygt og at pasientene gjenvinner sin respons (Rousselot et al., 2014). Man skal huske at disse pasientene er de mest lettbehandlede KML-tilfellene. Man har også sett at noen pasienter har detekterbar restsykdom opp til MR3 over lang tid uten å få tilbakefall, s.k. fluctuators. Dette er i analogi med erfaringer etter SCT og IFN. Tap av MR3 er derfor anbefalt som tidspunkt for rebehandling (Rousselot et al., 2014). Etter erfaringene av spesielt EuroSKI (imatinib-behandlede) der man har lett etter optimale cut-off foreslår man at man gjennomfører seponeringsforsøk har behandlet med TKI i 5 år og tentativt minst 2 år i MR4. Fra 3 års behandlingstid vinner man ca. 3 % flere relapsfrie pasienter per år man venter. Dette betyr at lege og pasient selv må velge hvor lenge man skal vente før seponeringsforsøk skal gjøres. Subjektive bivirkninger kan spille rolle for denne vurderingen. Vi anbefaler at pasienter som har vært resistente for TKI foreløpig ikke stoppes i klinisk praksis. Pasienter som var intolerante for første TKI og deretter fikk dyp repons på TKI nummer to kan stoppe som angitt ovenfor.
Tabell 10.2: Krav til å gjøre seponeringsforsøk (Hochhaus et al., 2020)
Resistens mot noen TKI før? | Nei |
Varighet av TKI-behandling | 5 år tentativt for imatinib og 4 år for 2TKI |
Varighet MR4 | 2 år |
God monitorering | PCR-lab må kunne gi svar innen 4 uker. PCR hver 6. uke i i 6 mnd, så hver annen måned til måned 12, så hver 3. mnd i 3 år, deretter hver 4.–6. mnd. Relaps etter 3 års oppfølging skjer nesten kun hos pasienter som har detekterbart transkript (og har i slike tilfeller meget langsom kinetikk (Richter et al Leukemia in transkript) |
Praktiske opplysninger
Transplantasjonsrelaterte problemstillinger: Søknad med problemstilling kan rettes til Norsk gruppe for allogene stamcelletransplantasjoner ved leder Tobias Gedde-Dahl, Seksjon for blodsykdommer, Medisinsk avdeling, OUS Rikshospitalet, 0027 Oslo.
DAStop2 | 2. gangs seponeringsforsøk av TKI hos pasienter som har gjennomført stoppforsøk som i EuroSKI studien. Pas byttes til dasatinib og kan stoppe dersom med MR4 i mer enn 1 år og de har fått total TKI-behandling i minimum 3 år fra 1. relaps. Info: mawa@sus.no eller regionansvarlig for KML. Inklusjon fra 2018, men pågår ennå |
SCAN-ALL | Studie av next generation sequencing (NGS) som sensitiv mutasjonsdeteksjon hos pasienter med KML og ikke-optimal respons respons. |
NordCML13/Labstop | Studie av immunologi og stamceller hos pasienter som skal stoppe TKI-behandling for TFR i vanlig klinisk praksis. Meget lovende avansert laboratoriemetodikk benyttes, og prøvetakingen er enkel. |
Bergen: | Bjørn Tore Gjertsen | bjorn.gjertsen@med.uib.no |
Oslo: | Tobias Gedde-Dahl | tgeddeda@ous-hf.no |
Stavanger: | Waleed Majeed | mawa@sus.no |
Tromsø: | Anders Vik | anders.vik@unn.no |
Trondheim: | Henrik Hjorth-Hansen | henrik.hjorth-hansen@ntnu.no |
Adresser for prøveforsendelse:
Cytogenetisk undersøkelse (karyotypering) og FISH. 10 mL perifert blod med heparin som anti-koagulant. Fra beinmarg tas 2–5 mL med tilsatt heparin i McCoys medium. Helst ikke på fredager eller dager før helligdager.
Seksjon for Cytogenetikk, Fagområde Kreftgenetikk
OUS-Montebello
N-0310 Oslo
Telefon: 23 93 44 37
Senter for medisinsk genetikk og molekylærmedisin
Haukeland Universitetssykehus
5021 Bergen
e-post: rhov@haukeland.no eller MGM@helse-bergen.no
Tel 55 97 54 75 Faks 55 97 54 79
Kvantitativ PCR og mutasjonsanalyse: 10 mL EDTA-blod sendes til:
OUS-Gaustad, Avdeling for patologi
Laboratorium for molekylærpatologi
PB 124 Blindern
0314 Oslo
Telefon: 23 07 87 82
e-post: signe.spetalen@ous-hf.no
Senter for medisinsk genetikk og molekylærmedisin
Haukeland Universitetssykehus
5021 Bergene-post: rhov@haukeland.no eller MGM@helse-bergen.no
Andre indolente lymfoproliferative sykdommer
Waldenströms makroglobulinemi
Sist faglig oppdatert: 23.12.2021
Innledning
Waldenströms makroglobulinemi (WM) defineres ved monoklonalt IgM i serum (uavhengig av konsentrasjon) og minst 10 % infiltrasjon av lymfoplasmacytisk lymfom i beinmarg (Castillo et al., 2020a; Leblond et al., 2017; Owen et al., 2003; Swerdlow et al., 2017). En mutasjon i MYD88-genet (MYD88 L265P) finnes i tumorcellene hos 90–95 % av pasientene, og mutert CXCR-4 forekommer hos ca. 1/3 (Castillo et al., 2019b; Treon et al., 2012).
WM oppdages oftest tilfeldig ved utredning av høy SR eller patologisk elektroforesefunn hos symptomfrie personer. Klinisk presentasjon kan være anemi, annen beinmargssvikt, hyperviskositet (eventuelt med retinopati) eller blødningstendens (Leblond et al., 2017). Perifer nevropati forekommer hos ca. 20 % og skyldes immunologisk effekt av det monoklonale immunglobulinet. Antistoffspesifisitet for myelin-assosiert glykoprotein (MAG) er en hyppig forekommende patogenetisk mekanisme for nevropati og den som er best kartlagt (D'Sa et al., 2017). Andre manifestasjoner som skyldes biologisk effekt av det monoklonale IgM forekommer hos mindre enn 5 % og omfatter kryoglobulinemi type I og II, autoimmun hemolytisk anemi, immun trombocytopeni, AL-amyloidose og akkvirert von Willebrands sykdom (Stone, 2011). Kuldeagglutininsykdom skilles i dag fra WM som en egen entitet (Randen et al., 2014). Bing-Neel-syndrom, direkte neoplastisk affeksjon av sentralnervesystemet (SNS), er sjelden og debuterer typisk 3–9 år etter diagnostisering av WM, men kan også inntre som første kliniske manifestasjon manifestasjon (Castillo et al., 2019a; Minnema et al., 2017). Medianoverlevelsen ved WM er mer enn 10 år (Leblond et al., 2017).
Diagnose
Når monoklonalt IgM er påvist, vil de viktigste differensialdiagnosene være andre B‑cellelymfomer (særlig marginalsonelymfom) og monoklonal gammopati uten funn av lymfom (enten uten kliniske manifestasjoner [MGUS] eller med klinisk sykdom til følge [monoclonal gammopathy of clinical significance, MGCS]) (Fermand et al., 2018).
Essensielle undersøkelser ved utredning
- Kapillær-elektroforese, evt. agarose-elektroforese med immunfiksasjon
- Kvantitering av immunglobulinklasser
- Beinmargsbiopsi
- Fokusert anamnese og klinisk undersøkelse mhp. lymfom og kliniske manifestasjoner av WM.
- Bildediagnostikk etter klinisk vurdering. Røntgen thorax og ultralyd abdomen er oftest tilstrekkelig.
- Hematologiske og klinisk-kjemiske blodprøver som ved andre lymfoproliferative sykdommer.
- Øyelegeundersøkelse ved synssymptomer, klinisk hyperviskositet eller IgM >40–50 g/L (Leblond et al., 2017).
- Ved mistanke om Bing-Neel-syndrom gir MR og spinalvæskeundersøkelse (inkludert væskestrømscytometri) nesten alltid diagnosen. Begge undersøkelsene må utføres, og i nevnte rekkefølge (Minnema et al., 2017). Differensialdiagnose: hyperviskositetssyndrom.
Eventuelle tilleggsundersøkelser
Påvisning av MYD88 L265P-mutasjon i beinmargsaspirat eller -biopsi kan gi nyttig informasjon i tvilstilfeller eller dersom behandling med ibrutinib planlegges (Castillo et al., 2020a; Treon et al., 2015b). Undersøkelse på mutasjon i CXCR-4-genet er i siste internasjonale konsensusdokument ikke anbefalt som klinisk rutine og er ikke rutinemessig tilgjengelig i Norge (Castillo et al., 2020a; Castillo et al., 2019b).
Behandling
Behandling av pasienter uten kliniske manifestasjoner er ikke vist å gi gevinst, og symptomfrie pasienter bør observeres ubehandlet inntil en behandlingsindikasjon inntrer (Leblond et al., 2017). Som regel er IgM-nivået i seg selv ikke indikasjon for behandling, men ved IgM >50–60 g/L må pasienten følges nøye med tanke på hyperviskositet. Noen anbefaler nå behandling ved IgM >60 g/L fordi behandlingstrengende manifestasjoner da vil melde seg raskt. Hos enkelte pasienter blir behandling nødvendig ved lavere IgM-nivåer. Behandling er i hovedsak indisert ved symptomgivende anemi eller annen klinisk relevant beinmargssvikt, hyperviskositet (med eller uten retinopati) eller klinisk blødningstendens (Castillo et al., 2020a; Leblond et al., 2017). Ved nevropati har nytten av klonalt rettet behandling vært omdiskutert, men en retrospektiv studie av MAG-assosiert nevropati tyder på effekt av kombinasjonsbehandling (Hospital et al., 2013). Internasjonalt anbefales nå WM-rettet behandling ved invalidiserende eller progredierende nevropati, særlig dersom MAG-antistoff påvises (D'Sa et al., 2017).
En rekke nyere kombinasjonsregimer gir høye responsrater og tilfredsstillende toleranse, se nedenfor. Monoterapi med klorambucil, fludarabin eller rituximab gir lave responsrater (30–50 %) og anbefales ikke i dag. Hos svært gamle og komorbide pasienter er klorambucil (helst i kombinasjon med rituximab) fortsatt et alternativ fordi det ofte tolereres godt (Leblond et al., 2013). Høydosebehandling med autolog stamcellestøtte (HMAS) er ikke anbefalt i første linje, men kan overveies i seinere behandlingslinjer hos selekterte, unge pasienter med klinisk aggressiv sykdom (Castillo et al., 2020a). Hos pasienter som seinere kan bli aktuelle for HMAS, bør man unngå purinanaloger og overveie stamcellehøsting under førstelinjebehandlingen (Castillo et al., 2020a).
Kombinasjonsbehandling med rituximab og bendamustin (R-benda) er i en randomisert studie vist å gi høye responstater og lang remisjonsvarighet og er nå et mye brukt førstelinjeregime (Castillo et al., 2020a; Rummel et al., 2013). R-benda er ikke sammenliknet med rituximab + syklofosfamid + deksametason (R-CD) i prospektive randomiserte studier, men retrospektive studier tyder på noe bedre effekt av R-benda (Castillo et al., 2020a). Bortezomib-baserte regimer gir rask effekt og høye responsrater (Dimopoulos et al., 2013). Pasienter med nevropati som får bortezomib, må overvåkes nøye mht. forverring av nevropatien, men god effekt på eksisterende nevropati er også observert (D'Sa et al., 2017). Bortezomib-basert behandling er særlig velegnet når raskt IgM-fall er viktig.
Pga. risiko for sekundær malignitet bør andre alternativer vurderes framfor fludarabin, særlig hos yngre pasienter. Ved hyperviskositet vil plasmaferese effektivt senke IgM-nivået og bedre de kliniske manifestasjonene (Leblond et al., 2017; Schwartz et al., 2016). Effekten er kortvarig, og medikamentell behandling må påbegynnes umiddelbart. Rituximab kan gi «IgM flare». Det er ikke et tegn på terapisvikt, men innebærer fare for hyperviskositet. Ved rituximab-holdige regimer hos pasienter med IgM >40–50 g/L, retinaforandringer eller klinisk hyperviskositet må rituximab derfor utgå fra første kur dersom man ikke starter med plasmaferese (Castillo et al., 2020a).
Behandling med Bruton tyrosinkinase(BTK)-hemmere har teoretisk et særlig sterkt rasjonale ved WM fordi MYD88 L265P-mutasjonen aktiverer B-cellereseptor-signalveien. Kliniske studier av BTK-hemmeren ibrutinib har bekreftet høye responsrater, lang remisjonsvarighet og høy sikkerhet (Dimopoulos et al., 2017; Treon et al., 2015a). I en ukontrollert, prospektiv studie fant man responsrater på 85–100 % hos tidligere behandlede pasienter (Treon et al., 2015a). Umutert MYD88-gen er assosiert med lavere responsrater og kortere remisjonsvarighet ved behandling med ibrutinib (Castillo et al., 2020a; Treon et al., 2015b). Sannsynligvis medfører også mutert CXCR-4 noe lavere responsrater (Castillo et al., 2020a; Castillo et al., 2019b). Ved avslutning av behandlingen er et seponeringssyndrom beskrevet hos 20 % av pasientene og en midlertidig økning av IgM-nivå hos 50 % (Castillo et al., 2020a).Ibrutinib er godkjent i Norge for annenlinjebehandling av WM. Nyere BTK-inhibitorer samt signalveihemmerne venetoklaks og idelalisib har også vist gode resultater (Castillo et al., 2020a), men er foreløpig ikke godkjent i Norge på indikasjonen WM.
Bing-Neel-syndromet har ved riktig behandling bedre prognose enn andre SNS-lymfomer. Høye responsrater og rimelig responsvarighet er rapportert både under behandling med ibrutinib og etter ulike typer kjemoimmunoterapi (se nedenfor) (Castillo et al., 2019a; Minnema et al., 2017). Høydose metotreksat eller intermediær- til høydose cytarabin er også effektivt, men anbefales ikke som førstelinjebehandling. Intratekal trippelbehandling kan gis som et tillegg etter individuell vurdering, men som eneste behandling har den ikke tilfredsstillende effekt (Minnema et al., 2017).
Behandling ved andre kliniske manifestasjoner er omtalt i annen litteratur (Castillo et al., 2020a; Leblond et al., 2017).
Behandlingsanbefalinger:
- Symptomfrie pasienter: Observere ubehandlet (Castillo et al., 2020a; Leblond et al., 2017).
- Behandlingstrengende pasienter: Rituximab-baserte kombinasjonsregimer foretrekkes framfor ren kjemoterapi dersom ikke kontraindisert (evidensgrad A) (Castillo et al., 2020a; Leblond et al., 2017).
- Førstelinjebehandling til pasienter med antatt middels til god behandlingstoleranse, med eller uten nevropati: R-benda (bendamustin-rituximab) i dosering omtalt nedenfor (evidensgrad B) (Castillo et al., 2020a; Rummel et al., 2013).
- Alternativt førstelinjeregime: BDR (bortezomib, deksametason og rituximab). BDR er særlig egnet dersom raskt IgM-fall er viktig eller ved dårlig beinmargsreserve, men bør vanligvis ikke gis til pasienter med nevropati. Dosering er omtalt nedenfor (evidensgrad C) (Castillo et al., 2020a; Dimopoulos et al., 2013; Leblond et al., 2017).
- Annenlinjebehandling ved terapisvikt:
- Alternativ 1, kjemoimmunoterapi: Det førstelinjeregimet pasienten ikke har fått tidligere, eller FC (fludarabin og cyklofosfamid), eller FCR (FC + rituximab) i dosering som ved KLL, eller R-CD (rituximab, cyklofosfamid og deksametason) i dosering angitt nedenfor (evidensgrad C) (Castillo et al., 2020.
- Alternativ 2: Ibrutinib (dosering omtalt nedenfor) (evidensgrad A) (Castillo et al., 2020a; Dimopoulos et al., 2017).
- Residivbehandling:
- Alternativ 1: Ved god respons på førstelinjebehandling og responsvarighet ≥2 år kan man gjenta førstelinjebehandlingen (evidensgrad C) (Castillo et al., 2020a; Leblond et al., 2017).
- Alternativ 2: Annenlinje kjemoimmunoterapi med et av regimene for terapisvikt i første linje (evidensgrad C) (Castillo et al., 2020a; Leblond et al., 2017).
- Alternativ 3: Ibrutinib (dosering angitt nedenfor) (evidensgrad A) (Castillo et al., 2020a; Dimopoulos et al., 2017; Treon et al., 2015a).
- Uttalt komorbide pasienter: Klorambucil (dosering som ved KLL), evt. kombinert med rituximab (evidensgrad C) (Leblond et al., 2013).
Behandlingsregimer
- R-benda (bendamustin og rituximab):
Bendamustin 90 mg/m2 dag 1 og 2.
Rituximab 375 mg/m2 i.v. dag
Ny syklus fra dag 28. - BDR (bortezomib, deksametason, rituximab):
- Initialt: Bortezomib 1,3 mg/m2 dag 1, 4, 8 og 11. Deksametason per os, 20 mg x 1 dag 1, 2, 4, 5, 8, 9, 11 og 12. Rituximab 375 mg/m2 i.v. dag 11. Ny syklus fra dag 22.
- Ved begynnende remisjon går man over til ukentlig bortezomib: Bortezomib 1,6 mg/m2 dag 1, 8 og 15. Deksametason per os, 20 mg dag 1, 2, 8, 9, 15 og 16. Rituximab 375 mg/m2 i.v. dag 15. Ny syklus fra dag 28.
- Ibrutinib: 420 mg x 1 per os. Dosereduksjon kan forsøkes ved bivirkninger.
- Deksametason 20 mg dag 1. Rituximab 375 mg/m2 i.v. dag 1.
Cyklofosfamid 100 mg/m2 x 2 per os dag 1–5, totalt 1000 mg/m2 per syklus.
Ny syklus fra dag 22. - Øvrige regimer: Se ovenfor.
Oppfølging
Under behandling: Generelle prinsipper for valgt behandlingsregime gjelder også ved WM. Effekt monitoreres først og fremst ved IgM-nivå og klinisk tilstand (Leblond et al., 2017). Ved kjemoimmunoterapi for WM kan effekten inntre mye langsommere enn ved andre lymfomer eller myelomatose, og generelt anbefales det derfor ikke å skifte regime før etter 2–3 sykluser (Castillo et al., 2020b). Beinmargsbiopsi overveies etter ½ år. Immunokjemoterapi seponeres ved oppnådd maksimal respons. IgM kan fortsette å synke i inntil et år, og noen legger inn en observasjonsperiode etter de første 4–6 kurene (Castillo et al., 2020b). Behandling med ibrutinib bør sannsynligvis fortsette til progresjon eller intoleranse (Castillo et al., 2020a).
I behandlingsfrie perioder: Klinisk tilstand og IgM-nivå, og for øvrig hematologiske og klinisk-kjemiske prøver som hos lymfompasienter. Årlig øyelegeundersøkelse ved IgM >40–50 g/L.
Behandlingsansvar: De fleste sykehus med hematologisk spesialkompetanse bør kunne utrede, kontrollere og behandle pasienter med WM. Ved problemer eller uvanlige situasjoner bør man rådføre seg med universitetssykehus eller hematolog med spesiell interesse for WM.
Hårcelleleukemi
Sist faglig oppdatert: 23.12.2021
Hårcelleleukemi er en langsomtvoksende B-celleproliferativ sykdom (Swerdlow et al., 2017). Tumorcellene, som infiltrerer beinmarg og milt, finnes oftest også i lav konsentrasjon i blod. Ved riktig behandling skiller overlevelsen seg sannsynligvis ikke fra tilsvarende aldersgruppe i normalbefolkningen (Ghanima et al., 2002). De vanligste kliniske presentasjonene er infeksjonstendens ledsaget av anemi, nøytropeni, trombocytopeni og/eller splenomegali. Tumorcellene i blod og beinmarg har et karakteristisk utseende med rund, oval eller nyreformet kjerne med relativt finkornet kromatinstruktur. Cytoplasmaet er blågrått og relativt rikelig med uskarp, «hårete» avgrensning. Beinmargen er ofte vanskelig å aspirere («dry tap»). I beinmargsbiopsi kan celleholdigheten være lav. Immunfenotypen av leukemicellene viser et karakteristisk mønster (Swerdlow et al., 2017). Cellene uttrykker tartratresistent sur fosfatase, som kan påvises cytokjemisk eller ved immunfenotyping. Det foreligger nesten alltid en mutasjon i genet for protein-kinasen BRAF (BRAF V600E). Den kan påvises ved PCR og bekrefter diagnosen, men undersøkelsen er kun indisert ved tvil om diagnosen etter væskestrømscytometrisk undersøkelse og beinmargsbiopsi.
Behandlingsindikasjon er symptomgivende sykdom, oftest pga. beinmargssvikt med hemoglobin <12 g/dL, nøytrofile <1 x 109/L og/eller trombocytter <100 x 109/L. Fallende tendens i blodverdiene styrker behandlingsindikasjonen. Ved klinisk infeksjonstendens kan det være en fordel å starte behandling før nøytrofile har falt under 0,5–1,0, fordi behandlingen senker tallet ytterligere og øker infeksjonsrisikoen forbigående.
- Førstelinjebehandling er cladribin 0,14 mg/kg/dag som en 1–2 timers iv-infusjon i 5 dager (evidensgrad B) (Juliusson et al., 1995; Sarvaria et al., 2016).
Denne administrasjonsmåten er terapeutisk likeverdig og mer praktisk enn den godkjente, som er 0,09 mg/kg/dag som kontinuerlig iv infusjon i 7 dager. Pentostatin, en annen purinanalog, er et likeverdig alternativ, men er uregistrert og lite brukt i Norge. Behandlingen er svært effektiv, og ca. 90 % oppnår komplett remisjon, oftest av flere års varighet. Median tid til ny behandling er >10 år.
Residivbehandling gis med samme behandlingsindikasjon som primærbehandlingen og vanligvis med samme regime (Sarvaria et al., 2016). Ved kort varighet av siste remisjon (<1 år) vil mange velge rituximab 375 mg/m2 i.v. (4 doser med en ukes mellomrom), som også har utmerket effekt. Alternativt kan man velge en annen purinanalog eller gi rituximab og purinanalog i kombinasjon (Else et al., 2011).
B-prolymfocyttleukemi (B-PLL)
Sist faglig oppdatert: 23.12.2021
B-PLL utgjør mindre enn 1 % av B-celleleukemiene. Tumorcellene er modne B-celler med utseende som prolymfocytter med rund eller oval kjerne med kondensert kromatin og tydelig nukleolus (Else et al., 2011). Kjerne/cytoplasma-ratio er lavere enn ved KLL. Prolymfocytter utgjør typisk >55 % av cellene i perifert blod. Betydelig lymfocytose og splenomegali er karakteristisk, men lymfeknutesvulst er ikke vanlig. Anemi, trombocytopeni og B-symptomer er vanlig. Median debutalder er 65–70 år. Immunfenotypen er karakteristisk og viser ekspresjon av sIgM+/– IgD med høyere intensitet enn ved KLL, CD19, CD20, CD22, CD79a og b, FMC7. CD5 og CD23 er vanligvis negativ. Del(17p) og p 53 mutasjoner ses hos ca. 50 % av pasientene. Differensialdiagnoser inkluderer T-PLL, KLL, mantelcellelymfom, follikulært lymfom, lymfoplasmacytisk lymfom og hårcelleleukemi.
B-PLL responderer relativt dårlig på behandling, og dette har sammenheng med at p 53‑signalveien, som konvensjonell kjemoterapi og i noen grad monoklonale antistoffer er avhengig av, er defekt. CHOP, cladribin, COP, fludarabin, rituximab har vært forsøkt. Median overlevelse er 30–50 måneder.
T-prolymfocyttleukemi (T-PLL)
Sist faglig oppdatert: 23.12.2021
T-PLL er en sjelden, posttymisk T-celleneoplasi. Sykdomsmanifestasjonene er svært like de som ses ved B-PLL, men affeksjon av serøse hinner (pleuravæske og ascites) og hudinfiltrater er karakteristisk (Else et al., 2011). Pasienter med ataxia teleangiectasia har betydelig økt insidens av T-PLL. Median debutalder er 65 år. T-PLL er en aggressiv sykdom med median overlevelse ca. 8 måneder.
Tumorcellene er små til middels store. Kjernen er rund, oval eller irregulær og har nukleol, og cytoplasmaet er basofilt. Hos 25 % ses en småcellet variant (tidligere omtalt som T-KLL). Hos 5 % er kjernen irregulær, «cerebriform». Uavhengig av kjerneform er det typisk med cytoplasmatiske utløpere, «blebs». Beinmargen er diffust infiltrert. Hudinfiltrasjonen affiserer ansikt, oftest periorbitalt. Purpura og ødem er vanlig. Biopsi viser perivaskulære evt diffuse dermale infiltrater. Immunfenotyping viser ekspresjon av CD52, CD2, CD3, og CD7. 60 % er CD4+/CD8-, 25 % CD4+/CD8+ og 15 % CD4-/CD8+. T-celle-reseptorgenene er klonalt rearrangert. Tumorcellene uttrykker onkogenet TCL1. Abnorme karyotyper er vanlige, og i ca. 90 % er kromosom 14 involvert. Differensialdiagnoser er B-PLL, mycosis fungoides, KLL, adult T-celle-leukemi/lymfom, hårcelleleukemi og T-LGL.
- Purinanaloger gir responsrate <50 %.
- Alemtuzumab monoterapi gir responsrate >50 % og totaloverlevelse ca. 15 måneder.
- Gode responser, dvs. komplett remisjon morfologisk, bør konsolideres med HMAS eller allogen stamcelletransplantasjon hos pasienter <65 år uten komorbiditet. Det gir muligheter for kurasjon hos noen få pasienter.
Storcellet granulær lymfocyttleukemi (LGL-leukemi)
Sist faglig oppdatert: 23.12.2021
Et klinisk sykdomsbilde karakterisert ved økt antall store lymfocytter med azurofile, cytoplasmatiske granula i blod og kronisk nøytropeni ble beskrevet i 1985. Molekylærgenetisk påvisning av klonalt rearrangert TcR gjør at dette oftest oppfattes som klonal lymfoproliferativ sykdom, men grensen til monoklonal T-celleproliferasjon av usikker klinisk betydning er uklar. Large granular lymphocyte (LGL)-leukemi benyttes gjerne som betegnelse, men storcellet granulær lymfocyttleukemi har fått et visst innpass på norsk. Immunfenotypisk undersøkelse avdekker to varianter; CD3+ (T-cellesykdom) og CD3- (NK-cellesykdom) (Else et al., 2011).
T-LGL-leukemi
Denne formen utgjør minst 85 % av tilfellene. Median alder ved diagnose er 60 år. Forløpet er ofte indolent, og en tredel av pasientene er uten symptomer ved diagnose. Symptomene er knyttet til cytopenier, oftest gjentatte luftveisinfeksjoner og hudinfeksjoner pga nøytropeni. Opportunistiske infeksjoner forekommer vanligvis ikke. Anemi i form av DAT-negativ hemolytisk anemi eller erytroaplasi er heller ikke uvanlig. Sykdommer med immunpatogenese, spesielt revmatoid artritt, er hyppig assosiert med T-LGL, og en stort andel av de som oppfyller de diagnostiske kriteriene for Feltys syndrom (revmatoid artritt, splenomegali og nøytropeni) har T-LGL-leukemi.
Diagnostikk
Funn av >0,2 x 109/L store granulære lymfocytter i blod er ofte assosiert med klonale T‑lymfocytter. Pasientene har som oftest ikke noen framtredende lymfocytose. Diagnosen baserer seg på karakteristisk klinikk, påvisning av en klonal T-cellepopulasjon med karakteristisk immunfenotype (CD2+,CD3+,CD7+,CD8+,CD57+) og interstitiell infiltrasjon av disse cellene i beinmargen. Klonalt rearrangert T-cellereseptor påvises alltid (Else et al., 2011).
Behandling
Den lymfoproliferative tilstanden per se trenger sjelden eller aldri behandling. Transformasjon til aggressivt lymfom er en raritet. Det er nesten uten unntak symptomer pga. de ledsagende cytopeniene som representerer behandlingsindikasjonen. Det foreligger ingen prospektive behandlingsstudier. I en britisk-norsk retrospektiv studie svarte 80 % av pasienter med symptomgivende nøytropeni meget godt på ciklosporin eller metotrexat, eventuelt kombinert med filgrastim, mens responsratene ved behandling med syklofosfamid var noe lavere (Osuji et al., 2006). En fransk studie viste brukbare responsrater ved de samme behandlingsalternativene, men her var forholdet omvendt (Moignet et al., 2014).
- Anbefalt behandling: Ciklosporin (medikamentfastende konsentrasjoner av ciklosporin 100–150 ng/mL) eller metotrexat (10 mg/m2 ukentlig), eventuelt kombinert med filgrastim initialt i 2–3 måneder (evidensgrad C).
Pasienter som ikke responderer på ciklosporin, kan respondere på metotrexat og omvendt. Syklofosfamid er aktuelt som annenlinjebehandling. Hos noen få pasienter som ikke har respondert på noen av disse alternativene, eventuelt i kombinasjon med G-CSF, kan pentostatin eller alemtuzumab forsøkes. Sykdomsmanifestasjonene residiverer vanligvis etter noen måneder, unntaksvis få år, når behandlingen med ciklosporin eller metotrexat seponeres.
NK-LGL-leukemi
Pasientene med NK-LGL-leukemi tenderer til å være yngre enn pasientene med T-LGL-leukemi, median alder i underkant av 40 år. Sykdommen har vanligvis en aggressiv klinisk fenotype med raskt tiltakende lymfocytose, anemi, trombocytopeni og organomegali. Lymfeknutesvulst er vanligvis ikke særlig framtredende. Tumorcellene har immunfenotype CD3‑CD4CD8+CD56+CD57-. Noen anbefalt og effektiv behandling foreligger ikke.
Myelodysplastisk syndrom/neoplasi (MDS) og kronisk myelomonocytt-leukemi (KMML)
Introduksjon (MDS)
Sist faglig oppdatert: 21.12.2023
Myelodysplastisk syndrom eller myelodysplastisk neoplasi (MDS)
MDS (myelodysplastisk syndrom/neoplasi) er en heterogen gruppe klonale neoplasier med utgangspunkt i en hematopoietisk stamcelle. MDS er karakterisert ved dysplastisk, ineffektiv hematopoiese som resulterer i en eller flere cytopenier og ved økt risiko for transformasjon til akutt myelogen leukemi (AML).
Prognosen kan variere fra mange års levetid til noen måneder hos dem med rask progresjon til AML (Arber et al., 2016).
Assosierte immunologiske manifestasjoner forekommer hos 10-25% (Mekinian et al., 2016).
Forekomst og etiologi
Insidensen er MDS for alle aldersgrupper ca. 4–5/100.000/år, men for pasienter ≥ 70 år minst 20/100.000/år. Median alder ved diagnosen er 77 år. Færre enn 10% er yngre enn 50 år. Det er noe høyere forekomst blant menn med unntak hvis lav blastandel og del(5q) (Sekeres et al., 2022).
Eksponering for cytostatika, benzen og radioaktiv stråling øker risikoen for MDS. Akvirert aplastisk anemi, forekomst av hematologisk kreft i nær familie samt hereditære sykdomer som Fanconi anemi, telomer sykdommer, Shwachman-Diamond syndrom og Diamond-Blackfan anemi er assosiert med økt risiko for MDS (Baliakas et al., 2019).
Diagnostikk, utredning og klasifikkasjonssystemer (MDS)
Sist faglig oppdatert: 21.12.2023
Diagnosen MDS:
- Blodtall: En eller flere cytopenier: Hb <13 g/dl [menn], Hb < 12 g/dl [kvinner], neutrofile granulocytter < 1.8x109/l, trombocytter < 150 x109/l
- Morfologi: Dysplasi (>10%) for hver rekke (erytropoiese, granulopoiese, trombopoiese) og andel blaster bør angis i blod/beinmargsutstryk. Benmargsbiopsi: Cellularitet, fibrose, andel CD34+ celler bør angis. Dysplasi i megakaryopoiesen.
- Eksklusjonskriterier, se liste under
- Genetiske forandringer: Cytogenetiske- og molekylærgenetiske forandringer bør analyseres hos alle pasienter der MDS diagnosen har behandlingsmessige konsekvenser, men etter at differensialdiagnoser som blant annet vitaminmangler er ekskludert.
Hovedpunkter ved diagnostikk og utredning av MDS:
- Anamnese. Se pkt. "Anamnese og klinisk undersøkelse"
- Blodprøver inkludert blodutstryk Se pkt. "Blodprøver"
- Benmargsundersøkelser.
For diagnosen MDS må disse årsaker til dysplasi/cytopeni være ekskludert:
- Vitamin B12-/folinsyre mangel
- Nylig cytostatikabehandling
- HIV/HCV/HBV/Parvovirus B19/CMV/EBV-infeksjon
- Anemi ved kronisk sykdom
- Autoimmun cytopeni
- Medikamentindusert cytopeni
- Kronisk leversykdom
- Alkoholoverforbruk
- Tungmetall-eksponering
- Aplastisk anemi, myelofibrose, PNH
- Annen kreftsykdom med beinmargsaffeksjon
Benmargsundersøkelser:
Benmargsaspirat til:
- Morfologisk undersøkelse (MGG+ jernfarging). Se pkt "Morfologisk undersøkelse"
- Immunfenotyping (flowcytometri) bør gjøres ved diagnose. Se pkt "Flowcytometrisk analyse (FM) ved utredning av MDS"
- Cytogenetisk undersøkelse, men først etter at vanlige differesialdiagnoser er ekskludert. Se pkt. "Genetiske analyser"
- Somatiske mutasjoner ved next generation sequencing (NGS), dvs. målrettet dypsekvensering med myeloid mutasjonspanel. Dette bør gjøres hvis MDS mistanken er sterk og kun hvis det forventes å få behandlingsmessige konsekvenser. Prøven bør tas før behandlingsstart med cytostatika.
Benmargsbiopsi bør tas ved diagnosetidspunkt for vurdering av cellularitet, fibrosegrad, grad av dysplasi i megakaryopoiesen og for å utelukke andre årsaker til cytopeni.
Benmargsbiopsi kan brukes til genetiske undersøkelser, se pkt "Genetiske analyser" hvis problematisk aspirasjon/»dry tap»
WHO 2016 (4. utgave) | WHO 2022 (5. utgave) | ICC 2022 |
|---|---|---|
MDS-RS (MDS med ringsideroblaster) |
MDS-SF3B1 (MDS med lav blastandel og SF3B1 mutasjon) |
MDS-SF3B1 (MDS med mutert SF3B1)
|
MDS-del(5q)
| MDS-del(5q)
| MDS-del(5q)
|
| MDS-biTP53 (MDS med biallelisk TP53 inaktivering) | MDS med mutert TP53 |
MDS -U
|
| MDS, UNS uten dysplasi |
MDS-SLD
|
| MDS-UNS-SLD
|
MDS-MLD
|
| MDS-UNS-MLD
|
| MDS-LB, UNS (< 5%)a (MDS med lav blastandel) |
|
| MDS-hypoplastisk
|
|
MDS-EB1 (5-9%)a
| MDS-IB1 (5-9%)a
| MDS-EB (5-9%)a
|
MDS-EB2 (10-19%)a
| MDS-IB2 (10-19%)a
| MDS/AML (10-19%)
|
| MDS med fibrose (5-19%) |
|
Rullepreparat bør alltid tas samtidig med benmargsbiopsi, men er spesielt viktig ved problematisk aspirasjon/»dry tap».
Diagnostiske kriterier
Klassifikasjon av ulike undergrupper av MDS
To MDS klassifikasjoner ble publisert i 2022: International Consensus Classification (ICC) og 5. utgaven av WHO klassifikasjonen (Arber et al., 2022; Khoury et al., 2022).
Sammenliknet med WHO 2016, er den vesentligste forandringen økt fokus på genetiske avvik (Falini et al., 2023; Zhang et al., 2022).
Blaster i BM/PB | Karyotype | Mutasjoner | |
|---|---|---|---|
MDS med definerende genetiske avvik |
| ||
MDS-del(5q)
| BM <5% og PB < 2% | del(5q) alene eller med tillegg av 1 avvik, unntatt -7/ del(7q) |
|
MDS –SF3B1
| BM <5% og PB < 2% | Ikke del(5q), -7 eller kompleks karyotype | SF3B1 |
MDS-biTP53 (MDS med biallelisk TP53 inaktivering) | BM < 20% og PB < 20% | Vanligvis kompleks | 2 eller flere TP53 mutasjoner (VAF >10%) eller 1 TP53 mutasjon med VAF >50%, eller 1 TP53 mutasjon og samtidig del(17p) eller cnLOH |
MDS, morfologisk definert |
|
|
|
MDS-LB, UNS (MDS med lav blastandel) | BM <5% og PB < 2% |
|
|
MDS-hypoplastiska | BM <5% og PB < 2% |
|
|
MDS- IB1 (MDS med økt blastandel) | BM 5-9% eller PB 2-4% |
|
|
MDS- IB2 | BM 10-19% eller PB 5-19% eller Auerstaver |
|
|
MDS med fibrose | BM 5-19% PB 2-19% |
|
|
aAndel blaster i benmargen er angitt i parentes
Tabell 1.1.b WHO 2022 klassifikasjon av myelodysplastisk neoplasi (MDS) (Khoury et al., 2022)
aVed definisjon: < 30% av normal benmargscellularitet ved alder <70 år, og <20% ved alder > 70 år.
BM; benmarg, PB; perifert blod, cnLOH; kopinøytralt tap av heterozygositet, UNS; uten nærmere spesifikasjon.
Undergruppe av MDS | Dysplasi (antall linjer) | Blaster i BM og PB | Karyotype** | Mutasjoner*** |
|---|---|---|---|---|
MDS med SF3B1 (MDS-SF3B1) | Typisk > 1* | BM < 5% PB < 2% | Ikke isolert del(5q), - 7/del(7q), abn3q26.2 eller kompleks karyotype | SF3B1 (>10% VAF) Ikke ymulti-hit TP53 eller RUNX1. |
MDS-del(5q) | Typisk >1* | BM < 5% PB < 2%c | del(5q) med inntil 1 avvik til, unntatt -7/del(7q) | Enhver mut, men ikke multi-hit TP53 |
MDS- uten dysplasi
| 0 | BM < 5% PB < 2%c | -7, del(7q) eller kompleks karyotype | Enhver mut., Ikke ymulti-hit TP53 eller SF3B1 (>10% VAF; (MDS-SF3B1)) |
MDS-UNS-SLD med en-linje dysplasi | 1 | BM < 5% PB < 2%c | Ikke MDS del(5q) | Enhver mut., Ikke ymulti--hit TP53, og ikke MDS-SF3B1 |
MDS-UNS-MLD med multi-linje dysplasi | > 2 | BM < 5% PB < 2%c | Ikke MDS del(5q) | Enhver mut., Ikke ymulti--hit TP53, og ikke MDS-SF3B1 |
MDS med excess blaster (MDS-EB)b | Typisk > 1*
| BM 5-9% PB 2-9%c | Alle | Enhver mut., Ikke ymulti-i-hit TP53 |
MDS/AML** | Typisk > 1
| BM 10-19% PB 10-19d | Alle med unntak av AML definerende**** | Ikke NPM1, bZIP CEBPA eller TP53 |
MDS med mutert TP53 |
|
|
|
|
MDS med mutert TP53 | Enhver | BM 0-9% PB 0-9% | Kompleks karyotyope ofte med tap av 17pf | yMulti-hit TP53 mutasjon, eller TP53 mutasjon (VAF >10%) og samtidig kompleks karyotype, ofte med tap av 17p |
MDS/AML med mutert TP53 | Enhver | BM: 10-19% PB: 10-19% |
| Enhver somatisk TP53 mutasjon (VAF>10%) |
Alle undergrupper har minst en cytopeni
BCR-ABL1 rearrangering, eller hvilken som helst rearrangering assosiert med myeloide/lymfoide neoplasier med eosinofili og tyrosinkinase gen-fusjoner, ekskluderer diagnosen MDS trass i cytopenier
b 2% blaster i PB (en gang) eller 1% blaster i PB ved 2 ulike anledninger gir mandat til diagnosen MDS-EB.
yMulti-hit TP53 er definert som:
2 distinkte TP53 mutasjoner (hver med VAF >10%) eller
en enkel TP53 mutasjon med enten: 1) del(17p), 2) VAF på >50%, eller 3) Kopi-nøytralt tap av heterozygositet (cnLOH) ved 17p TP53 locus.
**Myelodysplasi-relaterte cytogenetiske avvik: Kompleks karyotype og/eller del(5q)/t(5q)/ add(5q), - 7/del(7q), +8, del(12p)/t(12p)/ add(12p), i(17q),-17/add(17p)/del(17p), del(20q), og idic(X)(q13)
***Myelodysplasi-relaterte genmutasjoner: ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, ZRSR2
****AML definerende genetiske avvik: RUNX1-RUNX1T1, CBFB-MYH11, PML-RARA, APL med andre RARA rearrangeringer. KMT2A rearrangeringer. DEK-NUP214, inv(3)(q21.3;q26.2)/GATA2-MECOM, andre MECOM rearrangeringer. BCR-ABL, mutert NPM1, bZIP CEBPA mutasjon.
MDS med EB og blastandel >10-19%: defineres etter ICC ved forekomst av:
- MDS-definerende genetiske avvik (cytogenetiske endringer/mutasjoner) som MDS/AML.
- AML-definerende genetiske avvik som AML.
Anamnese og klinisk undersøkelse
- Tenk medfødt/arvelig sykdom hos alle pasienter < 50 år. Se pkt. "Medfødt/arvelig sykdom"
- Tidligere cytostatika- eller strålebehandling, alkoholoverforbruk, medikament bivirkning, spesiell yrkeseksponering
- Infeksjons-og eller blødningstendens
- Autoimmune symptomer (hypothyreose, rheumatoid artritt, polymyalgia rheumatika, pernisiøs anemi, autoimmun vaskulitt, inflammatorisk tarmsykdom og Sjögrens syndrom). Autoinflammatoriske manifestasjoner som Sweets syndrom og pyoderma gangrenosum kan være debutsymptom. Se eget avsnitt.
Klinisk undersøkelse
- Miltundersøkelse
- Pasienter < 50 år bør vurderes med tanke på medfødte sykdommer. Se pkt "Medfødt/arvelig sykdom"
- Ved telomersykdom/dyskeratosis kongenita (DK) forekommer karakteristiske negleforandringer, hypo-pigmentering, oral leukoplaki og lungeaffeksjon.
- Fanconi anemi og Blackfan Diamond anemi er assosiert med skjelettanomalier (tommelforandringer, radiusforandringer), kortvoksthet, lite hode og hudforandringer
- GATA-2 mutasjoner er assosiert med vorter, lymfødem, lungeforandringer, atypisk mykobakterieinfeksjoner, og monocytopeni.
Blodprøver
- Hb, MCV, MCH, MCHC, retikulocyttter, hvite med differensialtelling, trombocytter, blodutstryk.
- Ferritin, transferrinmetning. Folsyre, vitamin B12, homocystein og evt. metylmalonsyre (MMA), TSH og FT4
- ASAT, ALAT, ALP, LD, bilirubin, haptoglobin, DAT, Kreatinin, kalsium, Urat, INR
- immunglobuliner, proteinelektroforese, S-EPO,
- HBV, HCV, EBV CMV, HIV
- Evt fosfatidyletanol (PEth) ved mistanke om alkoholrelatert cytopeni
- Ved hypoplastisk MDS: Parvovirus-PCR, flowcytometri m.t.p. PNH, (HLA mtp DR15)
- Spesialprøver dersom mistanke om arvelig sykdom (Se pkt. "Medfødt/arvelig sykdom")
Morfologisk undersøkelse
I henhold til den 5. ugaven av WHO klassifikasjonen skal det telles 500 kjerneholdige celler i benmargen og 200 kjerneholdige celler i perifert blod (Khoury et al., 2022).
Den morfologiske klassifikasjonen av MDS baserer seg på
- Andel blaster i perifert blod og benmarg angis i prosent:
- Type og grad av dysplasi (angi om det er ≥10 %). Beskriv dysplasi, konferer nedenfor
- Prosentandel ringsideroblaster ≥5 % og <15 % eller ≥15 %.
- ≥10 % dysplastiske celler i den aktuelle myeloide rekken er anbefalt for betegnelsen dysplastiske karakteristika (blod/benmarg)
Dyserytropoiese
- Irregulære /lobulerte kjerner
- Nukleær “budding» (»kjerneknoppskyting»)
- Internukleær bridging (broer mellom kjernene) (intercytoplasmatisk bridge ses hos normale)
- Karyorrhexis (fragmentering av kjernen med frittliggende kromatinstrukturer utenfor kjernen)
- Multinukleære
- Megaloblastære endringer
- Ringsideroblaster
- Vakuolisering
Dysgranulopoiese
- Uvanlig små/store granulocytter
- Hyposegmentering (pseudo-Pelger-Huet celler)
- Hypersegmentering
- Hypo-/agranulering
- Auer-staver
Dystrombopoiese
- Mikro-/mononukleære megakaryocytter.
- Adskilte kjerner.
Flowcytometrisk analyse (FM) ved utredning av MDS
Flowcytometrisk analyse av beinmarg er et nyttig tilleggsverktøy ved diagnose og oppfølging av MDS (Porwit et al., 2023; Wang et al., 2023). Ingen markør ved flowcytometri er spesifikk for MDS, men kan understøtte MDS-diagnosen. Flowcytometrisk analyse kan påvise avvik fra normal hematopoiese ved endret ekspresjon av antigener, endrede lysspredningsegenskaper og endret sammensetning av subpopulasjoner (Kern et al., 2023). Analysen kan også i noen tilfeller bekrefte tilstedeværelse av neoplastiske mastceller ved mistanke om systemisk mastocytose med assosiert hematologisk neoplasi, selv om dette best vurderes i biopsi. Ved flowanalyse vurderes andel og fenotype på myeloide progenitorer, B-celleprogenitorer, og modningsmønstrene i myeloid, monocytoid og erytroid cellerekke. Megakaryocytopoiesen kan ikke rutinemessig vurderes ved flowanalyse siden megakaryocyttene er for store for deteksjon i rutineoppsett.
Forandringer sterkt assosiert med MDS (Porwit et al., 2023):
- Myeloide progenitor celler: Andel CD34+ celler > 3% vurdert ved flow er sterkt assosiert med MDS/ MPN (når AML er utelukket). Grundig analyse av CD34+ og CD117+ myeloide progenitor celler er spesielt viktig fordi flere uavhengige antigener kan ha avvikende uttrykk, inkl aberrant uttrykk av lymfoide markører (CD5, CD7, CD56), som er sterkt assosiert med MDS. Avvikende fenotype på myeloide progenitor celler kan imidlertid også sees ved MPN og sjeldnere etter behandling for andre tilstander
- Granulopoiese: Redusert relativt andel granulocytter, redusert granularitet og endret modningsmønster i granulopoesien med endret uttrykk av CD13/CD11b, CD13/CD16 og CD15/CD10 samt aberrant uttrykk av lymfoide antigener mm.
- Monocytopoiese: Nedregulering av monocytt-assosierte differensieringsmarkører med avvikende modningsmønster (CD11b, CD13, CD14, CD36, CD64), og lav andel modne monocytter (CD300e+). Uttrykk av lymfoide antigener (hyppigst CD56). Ikke sjeldent er HLA-DR nedregulert på monocyttene, og det kan være vanskelig å skille monocytt -og granulocyttlinjen. Tilsvarende avvik kan også sees ved KMML og er hyppigere ved denne diagnosen enn ved MDS.
- Erytropoiese: Oftest økt relativ andel erytroide forstadier. Redusert ekspresjon eller tap av CD71 og/ eller CD36 og endret andel celler som uttrykker CD117 (økt eller redusert).
Det er publisert mange scoringssystemer for MDS som inkluderer flowcytometriske forandringer. Ogata-score fokuserer på avvik i progenitor celler ved MDS. Det er vanlig å bruke cut off på 2, og dette gir sensitivitet på ca. 70 % og spesifisitet på ca. 80 %. Nyere scoringssystemer som også inkluderer flowcytometriske avvik i den erytroide linje, som «integrated MDS-FS score (iFs) gir imidlertid bedre sensitivitet og spesifisitet (Cremers et al., 2017; Oelschlaegel et al., 2023).
Det er viktig å merke seg at morfologiske blaster kan være CD34 negative, og dermed ikke det samme som blastandelen angitt som CD34+ ved flowcytometri. Summen av andel celler som er positive for CD34 og celler som er CD34 negative, men positive for CD117 og HLA-DR (begge populasjoner er flowcytometriske «blaster») korrelerer bedre med morfologiske vurdering (Sandes et al., 2013). Det er fortsatt blasttallet ved morfologisk bedømmelse av et teknisk adekvat benmargsaspirat (av erfaren morfolog) som skal brukes ved klassifisering av MDS.
Flowcytometri er spesielt verdifull ved utredning av pasienter med negativ cytogenetikk, uspsifikke mutasjoner og inkonklusiv morfologi, men skal ikke brukes alene til å diagnostisere MDS.
Benmargsprøvetaking til flowcytometri: Prøver bør analyseres innen 24 timer. Hvis dette ikke er mulig, bør prøven oppbevares ved romtemperatur. Oppsett i løpet av 36 -72 timer kan være akseptabelt, men er ikke ideelt og påvirker vurdering av lysspredningsegenskaper og andel døde celle.
Blod til flowcytometri: Ikke nødvendig ved diagnostikk av MDS.
Genetiske analyser
Generelt:
Kromosomale avvik påvises hos ca 50 % med nydiagnostisert MDS, somatiske genmutasjoner hos ca 90 %.
AML definerende avvik:
Følgende avvik er assosiert med AML og definerer diagnosen AML ved andel blaster < 20% : RUNX1T1, CBFB-MYH11, PML-RARA, APL med andre RARA rearrangeringer, KMT2A, DEK-NUP214, NUP98 rearrangeringer, inv(3)(q21.3;q26.2)/ GATA2-MECOM, andre MECOM rearrangeringer og mutert NPM1. (Arber et al., 2022; Khoury et al., 2022).
I WHO 2022 klassifiksjonen er det ikke krav til økt andel blaster i blod eller benmarg, mens iht ICC kreves >10% i beinmarg.
Rekvisisjoner til genetisk undersøkelser:
- Husk kliniske opplysninger og indikasjon for undersøkelsen.
- Husk informasjon hvis mistanke om eller kjent medfødt tilstand.
Cytogenetisk analyse
Indikasjoner:
- Ved utredning av pasienter med sterk mistanke om MDS
- Hos alle pasienter med MDS for prognostisk klassifisering i henhold til IPSS-R/ IPSS-M (tabellene 1.1.4a,b,c).
Foretrukket prøvemateriale:
- Benmarg.
- Ved «dry tap» kan en liten beinmargsbiopsi (ikke knus; genetikerne vil knuse selv) sendes på McCoy
Hvis det er flere blaster i perifert blod og «dry tap», kan blod sendes.
Mutasjonsscreening med dypsekvensering med myeloid mutasjonspanel (NGS)
Indikasjoner:
- Ved utredning av pasienter med sterk mistanke om MDS
- Hos alle pasienter med MDS for prognostisk klassifisering i henhold til IPSS-M dersom dette antas å få konsekvenser for oppfølging / behandling, inkludert alle potensielle transplantasjonskandidater
- Pasienter med del(5q) før lenalidomidbehandling samt under behandling.
- Ved medfødte tilstander som ved Fanconi anemi/ telomersykdom o.l. for påvisning av overgang mot MDS, viktig for avklaring vedrørende allo-HCT
Foretrukket prøvemateriale:
- Benmarg.
- Ved «dry tap» kan en liten beinmargsbiopsi sendes direkte til laboratoriet. Avtal prøveforsendelse med eget laboratorium
- Blod kan sendes ved mislykket benmargsprøve
Metode:
- Somatiske mutasjoner kan påvises med neste generasjons sekvensering (NGS)
- Undersøkelsen bør utføres ved bruk av NGS med sensivitet på minst 5 %.
- (IPSS-M har grense på >2%; dvs. vår sensitivitet vanligvis litt for dårlig).
Tolkningsutfordringer:
- Hvis påvist variant ikke er sikker patogen eller benign, klassifiseres den som usikker; «variant av usikker betydning» (VUS)
- For varianter med variant-allelfrekvens 40-60 % (nær heterozygot) eller > 90 % (nær homozygot er det vanskelig å avgjøre om det foreligger en somatisk eller en kimbanevariant uten å sammenlikne med ikke-hematopoietisk vev.
- Ervervede somatiske mutasjoner identisk med dem vi finner ved MDS ses hos friske personer uten cytopeni og kalles klonal hematopoiese av usikkert potensiale (CHIP)
Rekvisisjon:
- Husk kliniske opplysninger og indikasjon for undersøkelsen.
- Husk informasjon hvis mistanke om eller kjent medfødt tilstand.
Premalign klonal hematopoiese
CHIP - Klonal hematopoiese med usikkert potensiale: Minst en MDS relatert mutasjon med VAF ≥ 2 % (VAF ≥ 4 % i X-bundne gener hos menn). Ikke cytopeni. De hyppigst muterte genene er DNMT3A, TET2 og ASXL1 (Jaiswal et al., 2014).
CCUS - Klonal cytopeni av usikker betydning: CHIP med cytopeni og fravær av diagnostiske kriterier for myeloid neoplasi (Valent et al., 2017).
CHIP og CCUS oppfattes som premaligne tilstander som kan progrediere til myeloide neoplasier. Ved CHIP er risiko for malign utvikling ca. 0,5 – 1 % per år, mens risikoen er høyere ved CCUS. Risikoen for progresjon fra CHIP og CCUS til myeloid neoplasi øker med klonstørrelse (VAF >10%), antallet somatiske mutasjoner og spesielt nærvær av mutasjoner som: TP53,SF3B1, SRSF2, U2AF1, JAK2, RUNX1, ZRSR2, IDH1 og IDH2 og graden av cytopenier. Om pasienter med CHIP skal følges opp, diskuteres. Pasienter med CCUS bør ha oppfølgning og ASH Educational program 2021 anbefalte blodprøver med differensialtelling hver 3.-6 måned. Legebesøk hvert eller hvert 2. år. Benmargskontroll hvis tiltagende cytopenier (Osman, 2021).
Medfødt/arvelig sykdom
Medfødt/arvelig sykdom bør vurderes hos pasienter < 50 år
Kriteriene for pasienter < 50 år som bør testes:
A1: Pasienten har symptomer/tegn på arvelig sykdom som disponerer for myeloid neoplasme:
Langvarig trombocytopeni, immunsvikt, hyppige virusinfeksjoner/ vorter, tidlig gråhåret, annen cancersykdom
Anomalier som negledystrofi, munnslimhinneforandringer, hypopigmentering, faciale forandringer, cafe au lait flekker, eller uforklaring lever-, pancreas- eller lungeaffeksjon, kortvoksthet, mikrocephali, radiusdefekter.
A2: Familien:
Minst 2 familiemedlemmer (1. og 2. gradsslektninger) med MDS/AML, langvarig trombcytopeni eller symptomer/tegn på myeloid neoplasi og der en er diagnostisert før fylte 50 år.
A3: Familien:
En slektning med MDS/AML og minst 2 (1. eller 2. grads) slektninger med maligne tumores der minst en er diagnostisert før fylte 50 år.
A4: Familien:
> 3 slektninger (1. eller 2. grads slektninger) med myeloid sykdom, langvarig trombocytopeni eller symptomer/tegn på arvelig sykdom predisponerende for myeloid neoplasi uavhengig av deres alder
B: Somatisk mutasjonsanalyse kan av og til peke i retning av arvelige sykdomsgivende varianter. Spesielle gener som: RUNX1, GATA-2, TP53, ETV6, CEBPA og DDX41 kan ha både patogene somatiske og kimbanevarianter. Mistanken bør vekkes hvis variant allel frekvensen (VAF) av den spesifikke varianten er i nivå 40-60 % (nær heterozygot) eller > 90 % (nær homozygot).
DDX41 varianter synes å være en av de mest vanlige patogene kimbanevarianter sompredisponerer for MDS and AML hos voksne (forkomst 2-5%). Ulikt de andre variantene, oppdages disse gjerne i 55-60 års alderen
C: Pasienten < 50 år med MDS/AML, som ikke fyller kriteriene for A eller B, men har kromosom 7 aberrasjoner som monosomi 7, del(7q)/der(7)].
Diagnostikk av patogene kimbanevarianter:
Patogene kimbane varianter påvises lettest ved blodprøve (EDTA-blod) og/ eller ved prøve fra munnslimhinne. Ved medfødte tilstander som Fanconi anemi, telomersykdommer, Blackfan Diamond Anemi og lignende sykdommer er blodprøve tilstrekkelig, men ved patogene varianter som kan være både somatiske og kinbaneassosiert (varianter i RUNX1, DDX41, TP53, GATA-2) er det nødvendig med prøve fra et ikke-hemtopoietisk vev i tillegg til blod. Slimhinneprøve brukes ofte, men en må være sikker på at den ikke er kontaminert med blod (hvis f. eks. mucositt med blødning). Annet ikke-hematopoietisk vev kan brukes. Gull-standard er fibroblaster som er dyrket i kultur (fra hudbiopsi). Dette kan nå gjøres i Norge, men dyrkningen tar lang tid og er ikke ledd i rutinediagnostikk. Foreløpig bør det kun gjøres hvis det er usikkerhet knyttet til annet ikke-hematopoietisk vev.
Ved mistanke om kimbansemutasjon sendes EDTA-blod /slimhinne til et medisinsk genetisk laboratorium for undersøkelse. Se «Norsk portal for medisinsk genetiske analyser» for aktuelle laboratorier i Norge. Det fremkommer der hvilke genpaneler de enkelte avdelinger har og hvilken prøverevisisjon som bør brukes.
Husk også at for slimhinneprøver bruker ulike laboratorier forskjellige kit for analyse. Det aktuelle laboratoriet bør kontaktes før prøven tas, slik at prøven blir tatt på riktig måte.
Rekvisisjonen: Må inneholde adekvat problemstilling og familieanamnese slik at genetiker kan vurdere aktuelle gener for testing. Kontakt genetiker hvis usikkerhet.
Oppdagelse av patogene kimbane varianter er av vesentlig betydning for oppfølgning, behandling og ved seleksjon av donor ved allo-HCT.
Vurdering av benmargsfunn/ oppfølgning ved usikker MDS diagnose
Ved stabil cytopeni / dysplasi uten blastøkning og benmargs-ringsideroblaster < 5 % og uten cytogenetiske og molekylærgenetiske avvik, bør det tas et nytt benmargsaspirat for morfologisk vurdering etter (3-) 6 måneder. Ved tiltagende cytopeni, bør ny benmargsprøve tas tidligere.
Prognostiske scoringssystemer (MDS)
Sist faglig oppdatert: 21.12.2023
Prognose
MDS er en svært heterogen gruppe sykdommer med stor variasjon i overlevelse. Prognostiske verktøy basert på store pasientmaterialer kan forespeile forventet sykdomsutvikling (levetid, progresjon til AML) Det prognostiske verktøyet inndeler pasientene i ulike risikogrupper (lav, intermediær eller høy risiko).
IPSS-M er foreløpig ikke validert for behandling. Frem til mer erfaring med IPSS-M foreligger, anbefales at IPSS-R primært brukes med tanke på behandling. IPSS-M bør imidlertid også brukes idet det gir tilleggsinformasjon om patogene somatiske varianter av betydning for behandlingen og for å få mer erfaring med IPSS-M.Webkalkulator for IPSS-M, IPSS-R og aldersjustert IPSS-R: https://mds-risk-model.com/.
Viktige moment ved modellene og bruk av prognoseverktøy/kalkulator:
-Modellene er laget og validert for bruk ved diagnosetidspunkt, men kan antagelig gi nyttig prognostisk informsjon også etter oppstart av behandling eller ved utvikling av sykdommen.
-IPSS-R er utviklet kun for primær og ubehandlet MDS. IPSS-M er utviklet for både primær og sekundær/terapirelatert MDS.
-IPSS-R og IPSS-M bygger på cytogenetiske avvik og kliniske data som blastandel og cytopenier. ---IPSS-M bygger i tillegg på 31 molekylærgenetiske avvik/mutasjoner.
-IPSS-R kan alderskorrigeres. IPSS-M har alder integrert i score.
-IPSS-R har 5 risikokategorier: veldig lav, lav, intermediær, høy, veldig høy,
-IPSS-M har 6 risikokategorier: veldig lav, lav, moderat lav, moderat høy, høy, veldig høy.
Om molekylære data i IPSS-M:
For korrekt IPSS-M skår skal det for påvist mutasjon velges «yes», for ikke-påvist mutasjon velges «No», for ikke undersøkt mutasjon «Not Assessed». Ved mange manglende verdier blir risikoestimatet usikkert.
NPM1 tilstedeværelse tilsier at pasienten iht WHO 2022 automatisk skal klassifiseres som AML (se unntak under KMML). I ICC klassifikasjonen kreves et blasttall på minst 10% i tillegg for diagnosen AML. Mutasjoner med VAF > 2 % inkluderes i IPSS-M.
Prognostiske subgrupper | Cytogenetiske abnormaliteter | Median overlevelse (år) | Median tid til 25 % har transformert til AML | |||
|---|---|---|---|---|---|---|
Very good | -Y, del(11q) | 5.4 | Ikke nådd | |||
Good | Normal, del(5q), del(12p), del(20q), doble inkludert del(5q) | 4.8 | 9.4 | |||
Intermediate | Del(7q),+8,+19, i(17q), enhver annen enkel eller dobbel uavhengig klon | 2.7 | 2.5 | |||
Poor | -7, inv(3)/t(3q)/del(3q), 3 abnormiteter | 1.5 | 1.7 | |||
Very poor | Komplex: >3 abnormaliteter | 0.7 | 0.7 | |||
IPSS-R | Risiko score | Pasienter (andel) | Overlevelse (median, år) | AML transformasjon (25% av pasienter, år) 95% KI | |
|---|---|---|---|---|---|
Veldig lav | ≤1.5 | 19% | 8.8 | Ikke nådd (14.5-ikke oppnådd | |
Lav | >1.5 - 3 | 38% | 5.3 | 10.8 (9.2-ikke oppnådd) | |
Intermediær | >3 - 4.5 | 20% | 3.0 | 3.2 (2.8-4.4) | |
Høy | >4.5 - 6 | 13% | 1.6 | 1.4 (1.1-1.7) | |
Veldig høy | >6 | 10% | 0.8 | 0.73 (0.7-0.9) | |
IPSS-M
| Risiko score | Pasienter (andel) | Overlevelse (median, år)
| Overlevelse (25%-75% range, år) | Leukemifri overlevelse (median, år) | Leukemifri overlevelse (25%-75% range, år) |
|---|---|---|---|---|---|---|
Veldig lav | ≤ -1.5 | 14% | 10.6
| (5.1-17.4) | 9.7 | (5.0-17.4) |
Lav | >-1.5 - 0.5 | 33% | 6.0 | (3.0.-12.8) | 5.9 | (2.6.-12.0) |
Moderat lav | >-0.5 - 0 | 11% | 4.6 | (2.0-7.4) | 4.5 | (1.6-6.9) |
Moderat høy | >0 - 0.5 | 11% | 2.8
| (1.2-5.5) | 2.3 | (0.91-4.7) |
Veldig høy | >1.5 | 17% | 1.0 | (0.5-1.8) | 0.76 | (0.33-1.5) |
Om modellene
IPSS-R - Revidert Internasjonalt prognostisk scoring system.
- Revised International Prognostic Scoring System for MDS er basert på en register¬studie som inkluderte 7012 ubehandlede pasienter med MDS fra ulike europeiske land.
- Pasienter med sekundær/ behandlingsrelatert MDS og KMML med >12x109/L leukocytter
- ble ekskludert. IPSS-R tar utgangspunkt i blastandel i benmarg, cytogenetikk og grad av
- Cytopenier for å forutsi overlevelse og risiko for AML-utvikling. Dette gir 5 risikogrupper, men for praktiske formål deles MDS inn i 3 risiko¬grupper basert på IPSS-R:
- «Lav risiko» (Very Low og Low), «Intermediær risiko» og
«Høy risiko» (High og Very High). Verd å merke seg er at kompleks karyotype gir høyere risiko skår enn >10 % blaster i benmarg. IPSS-R kan aldersjusteres (IPSS-RA)
IPSS-M - Molekylær Internasjonalt prognostisk scoring system.
IPSS-M er en videreutvikling av IPSS-R som inkluderer mutasjoner i 31 MDS relaterte gener.
IPSS-M er utviklet for å gi en bedre prediksjon av totaloverlevelse, leukemifri overlevelse og risiko for transformasjon til AML. IPSS-M kan også forbedre risikovurdering med tanke på residiv og overlevelse etter allo-HCT og kan brukes ved både primær MDS og sekundær/terapirelatert MDS. IPSS-M er eksternt validert og viser bedre seperasjon mellom risikokategoriene sammenlignet med IPSS-R (Bernard et al., 2022).
- Mutert TP53 allel-status, en viktig prediktor for prognose og behandlingsrespons ved MDS (Bernard et al., 2020)
- Multi-hit TP53 er en samlebetegenlse for genetiske avvik som er assosiert med funksjonelt biallelisk tap av TP53. Multi-hit TP53 kan bekreftes ved tilstedeværelse av to eller flere distinkte TP53 mutasjoner med VAF > 10%, eller
- Én enkel TP53 mutasjon assosiert med enten:
- En cytogenetisk delesjon som involverer TP53 locus ved 17p13.1
- En VAF >50%, eller
- Kopinøytralt tap av heterozygositet (cnLOHved 17p TP53 locus (Bernard et al., 2020; Grob et al., 2022; Walter et al., 2021)
I fravær av LOH informasjon, vil tilstedværelse av en enkel TP53 mutasjon sammen med enhver kompleks karyotype bli ansett som likestilt med en multi-hit TP53 (Bernard et al., 2020; Grob et al., 2022).
- Ved både primær og sekundær MDS er prognosen etter behandling signifikant dårligere for pasienter med multi-hit TP53 mutasjons-status enn ved ved monoallelisk TP53 mutasjon. For å avgjøre om det foreligger multi-hit TP53 status, kreves spesialundersøkelser. Tilstedeværelse av kompleks karyotype og TP53 mutasjoner (multi-hit), er vist å predikere høy risiko for transformasjon til AML og død uavhengig av IPSS-R risiko skår, dårlig respons på kjemoterapi samt høy residivfrekvens etter allo-HCT (Bernard et al., 2020). Kompleks karyotype uten samtidig TP53 mutasjon (selv i nærvær av del(17p) kvalifiserer derimot ikke til denne kategorien, fordi dette ser ut til å ha noe bedre prognose enn TP53 –mutert MDS (Haase et al., 2019; Weinberg et al., 2022).
Prognose ved TP53 mutasjoner
- Multi-hit TP53 mutasjoner med eller uten kompleks karyotype: Disse pasientene har meget dårlig respons på intensiv kjemoterapi, høy recidivfrekvens inkludert etter allo-HCT og kort forventet overlevelse
- Monoalleliske TP53 mutasjoner med VAF >22%: Dårlig prognose, men bedre enn ved multi-hit TP53 mutasjoner.
- Mutert TP53 med VAF <10%: Samme prognose som ved villtype TP53.
Anbefaling:
Anbefaling for diagnose og prognostisk vurdering ved MDS:
- Alle pasienter bør klassifiseres i henhold til WHO 2022 og ICC 2022
- Alle pasienter bør risikostratifiseres i henhold til IPSS-R- og IPSS-M
- Multi-hit TP53 mutasjoner med eller uten kompleks karyotype har dårlig prognose, og svært høy recidivfrekvens etter kjemoterapi og allo-HCT
- Høyt transfusjonsbehov med jernakkumulering og komorbiditet har prognostisk ugunstig tilleggsverdi som bør vektlegges ved behandlingsvalg
- Raskt progredierende sykdom krever rask behandlingsstart og raskt-virkende medikamenter
Responsevaluering
Alle pasienter, men spesielt pasienter aktuelle for allo-HSC, bør vurderes etter respons kriterier fra «International Working Group» (IWG) av februar 2023, som erstatter kriteriene utgitt i 2006/2018 (Zeidan et al., 2023).
De oppdaterte kriteriene er ment å gi en bedre korrelasjon mellom pasientsentrerte resultater og kliniske studier i en tid med mange nye medikamenter.
Consensus proposal for revised International Working group respons criteria for higher risk MDS (Zeidan et al., 2023).
(IWG kriteriene) definerer fire aspekter av respons basert på behandlingsmål:
1. Endring i sykdommens naturlige forløp
2. Cytogenetisk respons
3. Hematologisk bedring (HI)
4. «Quality of life»
Kategori | Responskriterier (respons må vare i minst 4 uker) |
|---|---|
Komplett remisjon (CR) | Benmarg <5 % myeloblaster med normal modning av alle celleliner Dysplasi kan vedvare. Ikke transfundert med SAG eller plater, og ikke gitt EPO, TPO eller G-CSF de siste 2 ukene. Perifert blod: Hb ≥ 10 g/dl, Trombocytter ≥ 100 x109/L, Neutrofile ≥ 1.0 x109/L Blaster 0 |
Partiell remisjon (PR) | Alle CR kriterier med unntak av: Benmargsblaster redusert med ≥ 50 % relatert til før behandling, men fortsatt > 5 % Cellularitet ikke relevant |
Komplett remisjon (CR) med begrenset recovery av tellinger(CRL) CR en-linje (CRunl) CR bi-linje (CRbl) | Benmarg <5 % myeloblaster med normal modning av alle celleliner Dysplasi kan vedvare. Perifert blod: blaster 0. CRunl: Perifert blod: Møter ikke CR, men bare en av følgende: Hb>10 g/dl, trombocytter > 100x 109/L, Neutrofile ≥ 1.0 x109/L CRbl: Perifert blod: Møter ikke CR men bare 2 av følgende: Hb>10 g/dl, trombocytter > 100x 109/L, Neutrofile ≥ 1.0 x109/L |
Komplett remisjon med partiell hematologisk recovery (CRh) | Benmarg <5 % myeloblaster med normal modning av alle celleliner Dysplasi kan vedvare. Perifert blod: Møter ikke kriterier for CR og CRL. Ingen Hb grense nødvendig, plater >50 x 109/L, neutrofile >1,0 x 109/L, blaster 0%.. |
Hematologisk improvement (HI) | Hematologisk improvement definert på bakgrunn av IWG 2018 respons kriterier.
|
Overall respons rate (ORR) | ORR =CR (eller CR ekvivalent) + PR+CRL + CRh + HI. |
Ingen respons | Møter ikke kriterier for CR (eller CR ekvivalent), PR, CRL , CRh eller HI |
Ikke evaluerbar | Alle registrerte/randomiserte pasienter bør rapporteres i denominatoren av responsevaluerings-analyser, på linje med prinsippet intensjon om å behandle. Denne kategorien kan inkludere pasienter som enda ikke har fått responsevaluering, som lider en tidlig død, går ut av studiet tidlig, eller de med en teknisk suboptimal benmargsprøve som utelukker behandling. |
Cytogenetisk respons | Komplett: Bortfall av kromosomavvik uten tilkomst av nye. Partiell: > 50% reduksjon av kromosomavvik. |
Sykdomsprogresjon
| Oppfyller en av kriteriene under: ≥ 50 % blastøkning, og absolutt økning i blastandel med minst 5% fra før gjeldende behandlingslinje. Sykdomsprogresjon ved forverring av cytopenier: Gjentatt behov for transfusjon med enten SAG- erytrocytter eller plater innen 8 uker, ikke relatert til akutt interkurrent sykdom eller behandlingseffekt, i fravær av hematologisk forbedring i minst en blodlinje, som definert over. Progresjon til AML: > 50% økning i blaster fra utgangsvurdering til > 20% blaster |
Tilbakefall av sykdom | Oppfyller en av kriteriene under: Minst 5% benmargsblaster, og 50% økning fra tidligere målinger, eller utvikling av ekstramedullær sykdom (myeloid sarkom). Nedgang i blod-tellinger > 50% fra maks remisjon/responsnivå i Hb, trombocytter og absolutt reduksjon av Hb med 1,5 g/dl, til Hb < 10g/dl, eller trombocytter < 100 x109/L, eller neutrofile <1 x 109 /L, eller Repetert behov for røde blodceller eller trombocytt-transfusjoner, ikke relatert til akutt interkurrent sykdom, eller behandlngseffekter i fravær av hematologisk forbedring av minst en annen blodlinje. |
Pasient- rapportert utfall | Rapportering ved hjelp av et validert vurderingsverktøy. |
Behandling og oppfølging - sammendrag (MDS)
Sist faglig oppdatert: 21.12.2023
Oppfølgning av MDS pasient
- Regelmessig oppfølgning med klinisk vurdering og blodprøver
- Benmargsprøver gjentas ved mistanke om progresjon
- Enhver MDS-pasient i alle risiko-kategorier bør vurderes m.t.p. allo-HCT som er eneste kurative behandling, men komorbiditet, alder og ECOG må vektlegges
Behandlingsalgoritmer
Lav risiko- / intermediær risiko MDS
- Vurder kurativ behandling med allo-HCT ved intermediær risiko hos pasienter med tilleggsfaktorer som tilsier dårlig prognose og som ikke inngår i IPSS-R slik som: kromosomale monosomier, høyt transfusjonsbehov med jernakkumulering, alvorlig trombocytopeni med blødningstendens og ved tegn til sykdomsprogresjon. For mutasjonsvurdering, bruk IPSS-M der prognostisk ugunstige mutasjoner inngår.
- Vurder kurativ behandling med allo-HCT ved lav- risiko MDS: Bare unntaksvis aktuelt som ved trombocytopeni med blødningstendens eller svært transfusjonsbehov og jernakkumulering
- Ved anemi:
- Vurder indiksjon for ESA ± G-CSF hos pasienter med score 0 og 1 i.h.t den prediktive modellen for respons på ESA (Tabell 1.1.5.3)
- Vurder kombinasjonen ESA+/-G-CSF ved MDS med ringsideroblaster og/eller SF3B1. Synergistisk effekt mellom ESA+G-CSF gir meget høy responsrate.
Ved manglende effekt av G-CSF+ESA, er luspatercept aktuelt. - Vurder immunosuppressiv behandling som ATG og /eller cicklosporin hvis samtidig neutropeni og/eller trombocytopeni og hypo-/normocellulær benmarg med <5% blaster, ev. PNH klon og uten høy-risiko cytogenetikk hos pasient ≤ 70 år uten ringsideroblaster og fibrose
- Vurder lenalidomid hos pasienter med isolert del(5q) og blastandel <5% som er behandlingsrefraktære for ESA+/- G-CSF eller som ikke er aktuelle i henhold til den prediktive modellen (Tabell 1.1.5.3). Ved påvist mutert TP53, bør lenalidomid ikke gis.
- Gi blodtransfusjon for forbedring av symptomer, livskvalitet, komorbiditet. Pasientens ønske bør tas i betraktning framfor en universell Hb-grense ved transfusjon. Ikke indikasjon ved Hb verdier >12 g/dl
- Start jernchelerende behandling ved jernakkumulering (ved ferritin ≥ 1000 eller etter transfusjon av ca. 25 enheter blod). Spesielt viktig hvis allo-HCT kan bli aktuelt
- Vurder deltagelse i kliniske studier hvis alvorlig cytopeni, og transfusjonsbehov og svikt på konvensjonell behandling
- Følg opp pasient m.t.p. sykdomsprogresjon/ overgang til høyrisiko sykdom.
Høy risiko-MDS
Transplantasjonsaktuelle pasienter
- Kurativ behandling med allo-HCT hvis akseptabel komorbiditet
Ingen øvre aldersgrense, men sjelden >75 år - For transplantasjonsaktuell pasient, ikke vent med forberedende tiltak eller forbehandling
Aktuell behandling for pasienter som ikke er kandidat for allogen stamcelletransplantsjon
- AzacytidineMelphalan ved hypo-/normocellulær behmarg og normal cytogenetikk
- Pasientener bør tilbys deltagelse i klinisk studie hvis mulig.
Anemi
Bakgrunn
Rundt 90 % av pasienter med MDS har anemi. Mange har anemirelaterte symptomer som tilsier behov for behandling. For å redusere behovet for blodtransfusjon bør andre behandlingsalternativ som erytropoiese-stimulerende medisiner (ESAs) +/- G-CSF, lenalidomid og luspatercept vurderes.
Transfusjon av røde blodceller
For pasienter der allo-HCT ikke er aktuelt eller nært forestående, bør det tilstrebes et Hb-nivå som tar hensyn til komorbiditet og gir pasienten god livskvalitet. Pasientens Hb-ønske bør vektlegges opp til Hb verdier 11 til 12 g/dL (Nilsson-Ehle et al., 2011).
Behandling med erytropoiese-stimulerende legemidler (ESAs)
Behandling med ESAs kan øke Hb-nivået, redusere transfusjonsbehovet og øke livskvaliteten hos pasienter med lav – og intermediær risiko MDS (Nilsson-Ehle et al., 2011). Bivirkninger er sjeldne, men det er registrert hypertensjon og tromboembolisme (DVT, PE, hjerneslag) hos <5 % av pasientene. Risiko for tromboembolisme kan øke ved rask Hb-stigning, og det anbefales derfor hyppig kontroll ved behandlingsstart. Initialt bør Hb holdes < 12 g/dL. Tillegg av G-CSF kan ha en synergistisk effekt og kan indusere respons hos ESAs refraktære pasienter (Hellström-Lindberg et al., 2003). Bruk av ESAs har i retrospektive studier vist økt totaloverlevelse uten økt risiko for transformasjon til AML (Fenaux et al., 2018).
Indikasjon for ESAs+/- G-CSF
- Lav og intermediær risiko MDS
- Symptomatisk anemi, individuell vurdering, sjelden indisert ved Hb >10 g/dl.
- Prediktiv score for respons 0 eller 1 (Ikke ved score 2) (Hellström-Lindberg et al., 2003)
Eksklusjonskriterier
- Blaster ≥ 10 %
- Prediktiv score for respons score 2 (Hellström-Lindberg et al., 2003)
Hvis jernmangel er jernsubstitusjon nødvendig.
Transfusjonsbehov | Poeng | s-Epo | Poeng |
|---|---|---|---|
< 2 E SAG/ måned | 0 | < 500 U/L | 0 |
≥ 2 E SAG /måned | 1 | ≥ 500 U/L | 1 |
Predikted repons: 0 poeng 74 %, 1 poeng 23 %, 2 poeng 7 %
Kriterier for erytroid respons
Partiell erytroid respons
- Hos transfusjonsavhengige pasienter: Stabil anemi uten behov for transfusjoner
- Hos pasienter med stabil anemi: Økning i Hb ≥ 1.5 g/L
Komplett erytroid respons
- Stabil Hb >11.5 g/L
Dosering av erytropoiese-stimulerende medisiner
Behandlingsmål Hb <12 g/dl
Startfase:
Erytropoietin (EPO): Start med EPO 30 000 U/uke. Reduser initial dose ved nedsatt nyrefunksjon eller ved lav kroppsvekt. Øk til 30 000 U 2 ggr pr uke hvis manglende respons etter 8 ukers behandling. Doser >60 000 U/uke er ikke anbefalt.
Darbopoietin (DAR): Start med 300 µg/14 dgr eller 150 µg/uke. Reduser initial dose ved nedsatt nyrefunksjon eller ved lav kroppsvekt. Øk til 300 µg/uke hvis manglende effekt etter 8 uker. Unngå startdose 300 µg/uke, da dette kan gi rask og langvarig stigning i Hb med økt tromboserisiko.
G-CSF: Hvis manglende effekt av EPO/DAR etter 8 uker, vurder tilleggsbehandling med G-CSF (Jädersten et al., 2005). Start med 300 µg (30 MIE) eller ekvivalent dose en gang ukentlig, alternativt 120 µg (12 MIE) filgrastim 2–3 ggr ukentlig. Dosereduser ved stigning i ANC til 6–10 x 109/l. Maks dose: 300 µg (30 MIE) x 3 ukentlig. Evaluer effekt av behandling med G-CSF i tillegg til full dose ESA etter 8-(16) uker. Hvis manglende effekt, seponer ESA og G-CSF.
Langtidsvirkende G-CSF (pegfilgrastim) har ikke blitt evaluert ved MDS og anbefales ikke. Overdosering: Hvis Hb > øvre referanseområde, må behandlingen få en pause og venesectio vurderes. Doseredusert behandling kan restartes ved Hb < 12 g/dl.
Vedlikeholdsfase:
Ved komplett respons (se over), reduser dosen hver 8. uke eller øk intervallet til neste injeksjon (spesielt ved DAR). Median EPO-dose er 30 000 U/uke, selv om noen pasienter responderer på lavere dose; 5000–10 000 U/uke.
Monitorer ferritin regelmessig, vurder i.v/p.o jernsubstitusjon hvis ferritin faller under øvre referanseområde, spesielt ved symptomer på jernunderskudd (lav MCV).
Ved behandlingssvikt/ tap av respons:
- Vurder jern- eller vitamin B12-mangel
- Sjekk benmargen (aspirat) hvis ønsket behandlingseffekt ikke oppnås eller ved symptomer på sykdomsprogresjon.
Behandlingssvikt skyldes sykdomsprogresjon i 18–28 %.
MDS med ringsideroblaster (RS)/ SF3B1 og blaster <5% og transfusjonstrengende anemi
Indikasjonen for ESA +/- G-CSF er som ved MDS-SLD/MLD uten ringsideroblaster. Synergieffekten mellom ESA og G-CSF er imidlertid mer uttalt ved MDS-RS enn uten RS
Luspatercept er godkjent for MDS med ringsideroblaster med IPSS-R veldig lav, lav og intermediær risiko med transfusjontrengende anemi. Forutsetning for behandling er at pasientene ikke har effekt av, har mistet effekten av ESA+G-CSG eller at de ut fra den prediktive modellen ikke forventes å ha effekt av ESA (Tabell 11.6). Luspatercept gir økt grad av transfusjonsuavhengighet sammenlignet med placebo (henholdsvis 38% og 13% transfusjonuavhengighet i minst 8 uker) (Fenaux et al., 2020). Sekundære endepunktdata taler for mulig positiv effekt også med tanke på bedring av samtidig neutropeni og trombocytopeni, men grunnet få pasienter må effekten tolkes forsiktig (Garcia-Manero et al., 2022). Luspatercept er et rekombinant fusjonsprotein som binder TGF-beta superfamilie-ligander og reduserer signalering gjennom SMAD2/3. Dette resulterer i erytroid modning gjennom differensiering av sen-stadium erytrocyttforløpere (normoblaster) i benmargen.
Dosering av luspatercept er 1,0-1,75 mg/kg subkutant hver 3. uke. Anbefalt startdose 1,0 mg/kg, med økning til 1,33 mg/kg og deretter 1,75 mg/kg. Doseøkning til neste dosetrinn dersom ikke transfusjonsuavhengighet etter to påfølgende doser (6 uker) på aktuelle dosetrinn eller ved behov for nye transfusjoner etter oppnådd transfusjonsuavhengighet. I studien ble behandling avsluttet ved manglende effekt etter 25 uker (Fenaux et al., 2020).
Vanligste bivirkninger er fatigue, diare, asteni, kvalme, svimmelhet og ryggsmerter, oftest milde. Studien var ikke stor nok til sikkert å kunne vurdere om det foreligger økt risiko for transformasjon til AML, men det var ingen indikasjon på dette (Fenaux et al., 2020).
Behandling med luspatercept er besluttet ikke innført i Nye Metoder 13.12.2021 https://nyemetoder.no/metoder/luspatercept-reblozyl
Lenalidomid
Bakgrunn
Lenalidomid regnes som et immunmodulerende legemiddel som virker på E3 ubiquitin ligase cereblon, og induserer økt nedbryting av spesielle proteiner som er viktige for MDS -cellenes overlevelse. Virkningsmekanismen er fortsatt ikke fullstendig kartlagt (Fink et al., 2015).
Lenalidomid er godkjent for lav-risk MDS pasienter med 5q- med mangelende eller tapt respons på ESA eller for pasienter som ikke kan forventes å ha effekt av ESA+G-CSF etter den prediktive modellen for ESA behandling (Tabell 1.1.5.3).
Ved lav risiko MDS og del(5q) får ca. 76 % av pasientene redusert transfusjonsbehov og ca. 67 % blir transfusjonsuavhengige ved lenalidomid. Responsen er rask med median tid til effekt ca. 4-6 uker. Cytogenetisk respons er observert hos ca. 50 %. Responsen har en median varighet på ca. 2 år (Fenaux et al., 2011; Lian et al., 2016). Responsraten er høyere ved 10 mg/dag 21/28 dager sammenlignet med 5 mg/dag 21/28 dager (Fenaux et al., 2011). Mutert TP53 kan påvises hos 12-17 % av pasienter med lav risiko MDS med 5q- før behandling med lenalidomid, og klonen kan øke under lenalidomidbehandling. Forskjellige studier (om enn ikke alle) har vist lavere responsrate og lavere sannsynlighet for cytogenetisk remisjon hos pasienter med mutert TP53 enn hos dem med vill-type TP53. Det er i tillegg funnet kortere responsvarighet, høyere risiko for AML transformasjon, kortere event-free survival (EFS) og lavere overall survival (OS) hos pasienter med 5q- som har mutert TP53. Det tilrådes ikke oppstart med lenalidomid hos pasienter med 5q- hvis mutert TP53 er til stede. Under behandling med lenalidomid bør mutasjonsstatus sjekkes idet mutante TP53 kloner kan utvikle seg og ekspandere under lenalidomidbehandlingen (Jädersten et al., 2011; Saft et al., 2014). Disse muterte klonene kan ev. være uttrykk for sykdomsprogresjon. Således bør benmargen undersøkes for tilsynekomst av mutert TP53 under lenalidomidbehandling. Ved påvisning av mutert TP53 bør lenaliodmid seponeres.
Kandidater for allo-HCT kan behandles med lenalidomid. ;Mutert TP53 må ikke foreligge ved start, og pasientene må følges meget nøye mtp tilkomst av TP53 mutasjon. Benmargen bør sjekkes 3-4 måneder etter behandlingsstart, og videre hver 6. måned og hyppigere hvis nytilkomne cytopenier. Ved tilstedekomst av mutasjon i TP53, bør lenalidomid seponeres, og det bør klargjøres for allo-HCT. Også tilstedeværelse av TET2 og RUNX1 tilsier økt fare for sykdomsprogresjon.
Indikasjon for behandling lenalidomid
- Anemi hos pasienter med lav-risiko MDS med 5q- med tapt respons på ESAs +/- G-CSF eller lav sannsynlighet for respons iht. den prediktive modellen (se over).
Forsiktighet ved behandling med lenalidomid hvis
- Pasienter med ≥2 cytogenetiske avvik i tillegg til del(5q)
- Pasienter med kromosom 7 avvik (kontraindisert).
- Pasienter med påvist mutert TP53 (frarådes)
- Pasienter med blasttall >5 %
Dosering
Lenalidomid 10 mg gis x1 dag 1–21 med pause dag 22–28 (Fenaux et al., 2011). Hos eldre, svekkede pasienter og ved redusert nyrefunksjon (GFR 40–60ml/min) vurderes dosereduksjon.
Bivirkninger/varsomhet ved oppstart
Grad III og IV neutropeni eller trombocytopeni ses hos ca. 50 %. Ved start av behandling bør pasienten følges ukentlig. Ved uttalt neutropeni kan det gis G-CSF. Dosereduksjon kan bli nødvendig, men lavere dose kan gi redusert effekt, se over. Alle seksuelt aktive pasienter i fertil alder må bruke effektive prevensjonsmidler grunnet fare for alvorlige fosterskader. Menn må bruke kondom siden lenalidomid utskilles i sæd. Østrogenholdige prevensjonspreparater anbefales ikke grunnet økt risiko for VTE.
Kontroller
Første benmarg bør tas 3-4 mnd etter oppstart med lenalidomide og sjekkes for mutert TP53 og cytogenetisk remisjon. Hvis pasienten har oppnådd komplett cytogenetisk remisjon og mutert TP53 ikke foreligger, og pasientens neutrofile og trombocytter ikke har falt, kan neste benmargskontroll hos en pasient som ikke er aktuell for allo-HCT sjekkes etter 6-12 måneder.Hvis dette ikke er oppnådd, bør benmargen sjekkes to ganger årlig med analyse mtp tilkommet mutasjon i TP53 eller andre cytogenetiske avvik.
Ved tap av respons er det viktig å vurdere beinmargen med tanke på blastøkning, nytilkomne mutasjoner, eller nye cytogenetiske avvik. Det er økt risiko for progresjon til AML ved 2 eller flere cytogenetiske avvik i tillegg til del(5q). Hos disse pasientene må lenalidomid seponeres (Jädersten et al., 2011), og pasienten betraktes som høyrisikopasient.
Anbefaling:
- Lenalidomid er anbefalt til lav-risk MDS pasienter med 5q- med mangelende, tapt eller ikke forventet respons på ESAFor pasienter som behandles med lenalidomide skal det utføresanalyse mtp mutert TP53 og cytogenetisk remisjon etter 4 til 6 måneder. Ved cytogenetisk respons og fravær av TP53 mutasjon kontrolleres pasienten hver 6-12 måned, ellers to ganger årlig.
Immunsuppresjon (MDS)
Sist faglig oppdatert: 21.12.2023
Bakgrunn
Immunsupprimerende terapi (IST) med antithymocyttglobulin (ATG) og /eller Ciklosporin A (CsA) kan hos en liten gruppe MDS pasienter bedre cytopenier og redusere transfusjonsbehovet. IST har i kliniske studier gitt trilineære responser hos ca. 30 (16-67) % av pasientene (Haider et al., 2016; Platzbecker, 2019). Behandlingen kan gi langvarig effekt i en eller flere cellelinjer. For aplastisk anemi viste en randomisert studie at ATG fra hest (ATGAM) gav høyere respons rate enn ATG fra kanin. Tilsvarende er ikke gjort ved MDS. Tillegg av CsA til ATG har i en randomisert studie vist å gi høyere respons rate enn ATG alene ved aplastisk anemi. En retrospektiv studie har vist at tillegg av CsA til ATG økte response raten fra 27% til 51% ved MDS (Haider et al., 2016).
Faktorer som er assosiert med høy respons på IST: hypoplastisk benmarg, normal karyotype, forekomst av vevstype HLA-DR15, PNH klon og yngre pasienter uten benmargsfibrose. Pasienter >70 år bør ikke få ATG idet alvorlige bivirkninger da er observert.
Indikasjon for IST:
- MDS med IPSS-R very low / low/ intermediate med anemi og/ eller neutropeni og/ eller trombocytopeni
- Hypo- (normo-) blastisk benmarg med normal cytogenetikk, ev. med HLA DR 15 og PNH klon
Kontraindikasjon for IST:
- Tilstedeværelse av ringsideroblaster, mutasjon i SF3B1, benmargsfibrose og høy-risk cytogenetiske avvik (Stahl et al., 2018)
- >70 år Ikke bruk ATG, CsA kan brukes
Behandling med ATG
- ATG kan være forbundet med alvorlige bivirkninger som krever erfaring og ekspertise. Bør kun gis ved enheter der det er erfaring med ATG behandling.
- Bør ikke gis til pasienter >70 år eller ved vesentlig komorbiditet.
- ATG fra hest, ATGAM, er anbefalt ATG produkt
Hest ATG, Pfizer (ATGAM); 40 mg/kg, d 1–4) 1. valg. - Prednisolon (bør gis under ATG behandlingen). 1 mg/kg/dag på dag 1–10 (14) (trapp ned til seponering i løpet 14 dager) for å forhindre serumsyke (Kadia et al., 2012).
- Observasjon med tanke på serumsyke: utslett, artralgi/artritt og feber
- Profylakse mot Pneumocystis jirovechi og og aciclovir bør gis i minst 6 måneder.
- Ved uttalt og langvarig neutropeni anbefales sopp-profylakse mot aspergillus
- CsA gis i kombinasjon med ATG
- Hos pasienter >70 år eller der pasienten ikke forventes å tolerere ATG gis kun Ciclosporin.
- Ciclosporin A (CsA) oralt 2.5 mg/kg x2. Dosejustering etter serumspeil
- Ønsket CsA-speil 200 + /– 50 microgram/L. Obs. kreatininstigning, tremor, hypertensjon
- Profylakse mot Pneumocystis jirovechi bør gis i minst 6 måneder.
Responsevaluering
- Respons inntrer ofte først etter 3–8 måneder
- Respons kan være stabil. > 10 års varighet er rapportert
- Ved progressiv sykdom etter 3–6 måneder eller manglende respons etter 6–8 måneder, vurder annen behandling
- Hvis manglende respons etter 8 måneder, stopp CsA.
- Hvis minimal respons, fortsett til 12 måneder. CsA bør trappes langsomt ned.
Jernchelering (MDS)
Sist faglig oppdatert: 21.12.2023
Bakgrunn
Mange pasienter med MDS er avhengige av regelmessig blodtransfusjoner. Hver enhet blod inneholder 250 mg jern, og pasientene vil derfor over tid akkumulere store mengder jern. Målet med jernchelering er å forhindre toxisk jernavleiring i organer som hjerte og lever.
Det er 3 ulike jernchelerende medikamenter tilgjengelig i Norge; deferoksamin for intravenøst eller subkutant bruk og to per orale medikamenter: desferasirox og deferiprone. To store prospektive fase 2 studer er gjort med desferasirox hos MDS pasienter. 341 MDS pasienter ble behandlet med desferasirox i ett år i den ene og i TELESTO studien ble 225 pasienter randomisert 2:1 mot placebo (Gattermann et al., 2010). I begge studier ble Ferritinnivået og labilt plasmajern redusert. I TELESTO-studien økte event free survival (EFS) med 0.9 år fra 3 til 3.9 år i behandlingsarmen sammenlignet med placebo. . Det er gjort to prospektive observasjonsstudier fra et europeisk og fra et kanadisk register der begge viser forbedret overlevelse. Ved alvorlig jernoverbelastning og utilstrekkelig effekt av deferoksamin, kan kombinasjon med deferiprone eller deferasirox i vanlige doser prøves.
Jernoverbelastning før transplantasjon er assosiert med økt risiko for inferior post-transplant morbiditet og overlevelse (Armand et al., 2011). Transplantasjonsaktuelle MDS pasienter bør derfor starte jernchelering tidlig, gjerne ved Ferritin >800.
Indikasjon for jernchelering
- Pasienter med forventet langvarig blodtransfusjonsbehandling (MDS- veldig lav-, lav- og intermedær risiko ved ferritin ≥ 1000
- Pasienter aktuelle for allo-HCT ved ferritin ≥ 800- 1000.
Monitorering av jernchelering
Mål: Ferritin <1000 mg/L
Orale chelatorer
Deferasirox behandling
- Startdose 7–14 mg/kg. Vanlig vedlikeholdsdose: 14–28 mg/kg. Max-dose 28 mg/kg.
- Bør unngås hvis nyresvikt.
- Kontroll etter oppstart: Ukentlig de første 4 uker, så månedlig: Kreatinin, ASAT, ALAT
Hvis kreatinin >2ULN, bør deferasirox stoppes. Sjekk urin: Obs. nefritt. Hvis restart, bør dosen reduseres
Deferiprone behandling
- Deferiprone 75 mg/kg (fordelt på 3 doser)
- En liten andel blir neutropene, agranulocytose er beskrevet (meget sjelden).
- Kontroll etter oppstart: Blodtall ukentlig initialt
- Anbefales ikke til neutropene pasienter
- Kan kombineres med deferoksamin for bedre jernchelering
Anbefaling:
- Jernchelering anbefales til
- A. Pasienter med forventet langvarig blodtransfusjonsbehandling (MDS- veldig lav-, lav- og intermedær risiko ved ferritin ≥ 1000
- B, Pasienter aktuelle for allo-HCT ved ferritin ≥ 800- 1000.
Parenterale chelatorer
Deferoksamin (behandling)
- Vitamin C 2–3 mg/kg/d (bedre jernutskillelsen). Bør starte 4 uker etter oppstart av deferoksamin. Obs. høyere dose med vit. C er assosiert med hjertearrytmier.
- Deferoksamin 40 mg/kg (20–50 mg) ved subcutan infusjon over 8–12 timer 5–7 dager i uken.
- Alternativt: 5–10 g kontinuerlig over 5 dager i veneport.
- Kontinuering 24 timers (uavbrutt) infusjon med deferoksamin bør vurderes ved ferritin persisterende >2500 og hjertesykdom. Kombinasjonabehandling med deferiprone eller deferasirox bør da vurderes
Trombocytopeni (MDS)
Sist faglig oppdatert: 21.12.2023
Trombocytopeni er observert hos 40–65 % av pasienter med MDS. Hos 12 % av alle MDS- pasienter er blødninger primær dødsårsak (Basood et al., 2018). Behandlingsmulighetene er begrenset. Noen MDS-pasienter kan ha funksjonelle trombocyttdefekter.
Trombocytt transfusjoner
Platetransfusjoner kan i handlingsøyemed gis for å stoppe blødninger, eller profylaktisk før invasive prosedyrer. Ulempene er risiko for transfusjonsreaksjoner og alloimmunisering. Alloimmunisering medfører behov for HLA-forlikelig trombocyttkonsentrat.
Anbefaling:
- Gi platetransfusjon til trombocytopene pasienter ved moderat eller alvorlig blødning (nærmest uavhengig av trombocyttall). Noen få pasienter trenger trombocytter ved relativt høye trombocytt-tall pga. platedysfunksjon.
- Profylaktisk platetransfusjon ved trombocytter under et fastlagt nivå anbefales vanligvis ikke ved kronisk trombocytopeni hvis pasienten ikke blør.
- Transexamsyre (Cyklokapron) 500–1000 mg 3–4 ganger daglig kan gis til trombocytopene pasienter med blødningstendens. Forsiktig hvis hematuri (Antun et al., 2013).
Trombopoietin reseptoragonister
Flere kliniske studier har undersøkt de to TPO-reseptoragonistene romiplostim og eltrombopag mot placebo (Meng et al., 2020). Det er vist mindre blødningstendens ved behandling med TPO-reseptoragonistene. Det har vært bekymring rundt sikkerheten til medikamentene da noen studier med romiplostim har vist blastøkning og tilfeller med progresjon til AML. Studier har ev. vist litt mindre risiko ved eltrombopag (Oliva et al., 2017; Wang et al., 2022). Det er lite eller ingen evidens for forskjeller i mortalitet ved bruk av TPO-agonister vs placebo. TPO-agonister synes å gi mindre blødningstendens. Det er fortsatt uklart om TPO-agonister gir økt risiko for transformasjon til AML.
Anbefaling:
- Verken eltrombopag eller romiplostim har MDS som indikasjon. Anbefales fortrinnsvis brukt i studier inntil sikrere data foreligger.
- Hvis Tpo agonist skal forsøkes, må blasttallet i benmargen være < 5%. Pasient og lege må være oppmerksomme på mulig blastøkning
Neutropeni og infeksjonsprofylakse (MDS)
Sist faglig oppdatert: 21.12.2023
Injeksjoner med granulocytt-stimulerende faktor (G-CSF) kan vurderes profylaktisk til pasienter med neutropeni og recidiverende alvorlige infeksjoner. Publiserte data er begrensede. Det kan også vurderes å gi dette sammen med azacitidin behandling ved alvorlig neutropeni. Langtidsvirkende G-CSF har ikke blitt evaluert ved MDS og anbefales ikke.
Allogen stamcelletransplantasjon (allo-HCT) ved MDS
Sist faglig oppdatert: 21.12.2023
Bakgrunn
Allo-HCT er eneste kurative behandling ved MDS. Studier viser at allo-HCT gir klar overlevelsesgevinst ved høy-risk MDS sammenlignet med annen behandling (Kröger et al., 2021). Etter at vi i Norge innførte treosulfan/fludarabin som forbehandlingsregime, har fått bedre infeksjonsbehandling og forbedret HLA-diagnostikk, er resultatene for allo-HCT bedre enn tidligere (Vo et al., 2023). Dette sammen med bedre tilgang på ubeslektede givere, gjør at flere og eldre pasienter kan tilbys allo-HCT.
En forutsetning for å opprettholde en forsvarlig transplantasjonsrelatert dødelighet er at den enkelte pasientes komorbiditet, fysiske tilstand, motivasjon og kompliance vurderes nøye. Det er ikke lenger noen øvre aldersgense, men pasienter eldre enn 75 år er sjelden aktuelle. Utfallet etter allo-HCT kobles til:
- Sykdomsrelatert faktorer som andel blaster i benmarg og blod, type og antall kromosomavvik og mutasjoner (IPSS-R og IPSS-M)
- Pasientrelaterte faktorer der samtidig forekomst av andre sykdommer er av vesentlig betydning (Sorror, 2013) (HCT-CI, EBMT score og RIC score; beskrevet i AML kapitlet)
- Transplantasjonsrelaterte faktorer som kondisjoneringsregimet og HLA forlikelighet (Vittayawacharin et al., 2023)
I en eldre, større registerstudie fra EBMT fra 2000-2016 var «overall survival» (OS) 40%, Transplantasjonsrelatert dødelighet ca. 30% og recidiv-relatert død ca. 30% (Shimoni et al., 2021). Forbedret infeksjonsbehandling og metodikk for HLA typing samt bedre behandlingsstategier har gitt gunstigere resultater, konferer den randomiserte studien til Beelen et al. der 3-års overlevelse var 67% og 56% respektivt, beroende på kondisjoneringen (Beelen et al., 2022).
Pasienter aktuelle for allo-HCT
Anbefaling:
- Ta tidlig stilling til om det foreligger indikasjon for allo-HCT
- Vurder sykdommens prognose opp mot komorbiditet
- Hos pasient aktuell for allo-HCT: HLA-typ pasienten og start søk etter stamcellegiver
- Hvis mistanke om arvelig tilstand, bør genetisk utredning starte snarest
- Gi forebyggende jernchelering til pasienter som risikerer hemosiderose pga. høyt transfusjonsbehov.
Indikasjon for allo-HCT
- IPSS-R høy- og veldig høy risiko
- IPSS-R intermediær risiko hvis andre høy-risiko faktorer også forekommer slik som: høy-risiko mutasjoner (bruk IPSS-M), monosomier, alvorlige cytopenier, stort transfusjonsbehov eller sykdomsprogresjon
- Unge pasienter med IPSS-R lav risiko sykdom er unntaksvis aktuelle slik som ved alvorlig trombocytopeni med blødning eller svært transfusjonsbehov med hemosideroseutvikling eller hvis tegn til sykdomsprogresjon
- Endelig beslutning om allo-HCT tas etter avklaring av: MDS-risiko, donorstatus og pasientens komorbiditet
- Pasienter med kompleks karyotype og/ eller multiallelisk TP53 mutasjon har svært dårlig prognose med høy recidivfrekvens etter allo-HCT. Slike pasienter må vurderes nøye om de realistisk sett vil ha ha nytte av allo-HCT. De må informeres om dette.
- IPSS-M er ennå ikke validert i større transplantasjonsstudier, men anbefales i risikobedømmelsen og slik at erfaring erverves.
Cytoreduktiv behandling før allo-HCT
Kunnskapen om sykdomsmodifiserende behandling før allo-HCT er begrenset. Det foreligger mange studier. Noen peker på at lavere blasttall før transplantasjon gir bedre overlevelse, men dataene rundt pretransplantasjonsdebulking er fortsatt ikke entydige (Robin et al., 2015; Yahng et al., 2017). Internasjonalt ekspertpanel (2017) anbefalte blastreduksjon til <10% før allo-HCT spesielt ved non-myeoablativ kondisjonering (de Witte et al., 2017). Blastreduserende behandling er av større betydning ved redusert kondisjonering og aggressiv sykdom pga faren for recidiv i tidlig posttranssplantasjonsperiode, dvs. innen graft-versus –leukemia (GvL) effekten rekker å inntre.
Det er i Norge enighet om å gi cytoreduktive behandling til pasienter med:
- blastandel >10 %
- ved lavere blasttall enn 10% hvis raskt progrediserende eller svært høy risiko sykdom med forventet progresjon innen allo-HCT kan gjennomføres
- Allo-HCT bør ikke forsinkes av cytoreduktiv behandling hvis det raskt kan gjøres allo-HCT
Det er ikke vist forskjell mellom hypomethylerende midler og intensiv kjemoterapi (Damaj et al., 2012; Gerds et al., 2012; Vittayawacharin et al., 2023). Behandlingsvalg er avhengig av pasientens alder, comobiditet og cytogenetiske/molekylærgenetiske karakteristika. Dynamikken i sykdommen er viktig for behandlingsvalget. Det bør tilstrebes å gi så få behandlinger med kjemoterapi som mulig innen allo-HCT.
Det må sendes søknad til Nasjonal gruppe for allo-HCT (OUS) for pasienter aktuelle for allo-HCT.
Hypometylerende behandling (MDS)
Sist faglig oppdatert: 21.12.2023
Bakgrunn
Azacitidin anses å ha en hypometylerende effekt og således effekt på cancercellenes genuttrykk, men den eksakte virkningsmekanismen in vivo er fortsatt uavklart (Jones et al., 1980). Både OS og transformasjon til AML for pasienter med høy-risk MDS er vist forlenget og livskvaliteten forbedret i den azacitidin behandlede gruppen ved sammenligning med støttebehandling og AML induksjonskur. Nytten av azacitidin sammenlignet med støttebehandling er også vist for pasienter >75 år (Seymour et al., 2010). Overlevelsesgevinst er kun vist ved dosering 75 mg/m2 sc dag 1–7 i 28-dagers sykluser. Av praktiske hensyn brukes ofte dosering 100 mg/m2 sc dag 1–5 i 28 dagers sykluser, alternativt 75 mg/m2 dag 1–5 og dag 8–9, men det er ingen studier som har sammenlignet dette direkte med den opprinnelige doseringen. Flere studier viser behandlingseffekt av azacitidin på 40–50 % (komplett, partiell eller hematologisk bedring). Responsen er oftest kortvarig med median responsvarighet på ca. 1 år (Platzbecker, 2019). 91% har effekt innen 6 cykluser. En 48% responsrate innen 12 cykluser viser betydningen av ikke å avbryte behandlingen (hvis medikamentet tolereres) før i hvert fall 6 sykluser er gitt (Silverman et al., 2011).
Indikasjon for azacitidin
- MDS med høy og veldig høy risiko som bro til allo-HCT eller som eneste behandling
- MDS med intermediær risiko – særlig hvis IPSS-R er >3 og med høyrisiko-tilleggs-faktorer (benmargsfibrose, jernakkumulering, høy-risiko cytogenetiske avvik og høy-risiko mutasjoner) som eneste behandling eller som bro til allo-HCT
- Kun ved forventet levetid > 3 måneder.
Behandling med azacitidin
Dosering: Azacitidin 75 mg/m2 sc dag 1–7 hver 28. dag eller azacitidin 75 mg/m2 sc dag 1–5 og dag 8–9 hver 28. dag eller azacitidin 100 mg/m2 dag 1–5 hver 28. dag
Det trengs vanligvis 4–6 behandlingskurer for å oppnå effekt. Dersom progresjon ikke mistenkes, bør responsevaluering med benmargsaspirat gjøres etter 6 behandlingskurer.
Ved tvil om behandlingsrespons, vent 4-6 uker etter oppstart av siste kur før ny benmargsvurdering. Støttebehandling med G-CSF og profylaktisk antibiotika kan vurderes ved nøytropene infeksjoner. Ved respons, men økende cytopenier eller dårlig toleranse, kan doseintervall økes til 5–6 uker, alternativt kan dosen reduseres. De første to cyklusene bør gis med full dose. Behandlingen bør fortsette til progresjon av sykdom dersom pasienten tåler denne. Hvis behandlingen stopper, må recidiv forventes.
Ved effekt av azacitidin, er forventet responstid 6–24 måneder. Når azacitidinbehandlingen svikter, er prognosen dårlig med median overlevelse 4-6 måneder (Overvei da deltagelse i kliniske studier med nye medikamenter (Jabbour et al., 2015; Prébet et al., 2011).
AML-lignende kjemoterapi (MDS)
Sist faglig oppdatert: 21.12.2023
Bakgrunn
Tallrike studier med intensiv kjemoterapi, få randomiserte, er ved høy-risk MDS vist å gi høyere sannsynlighet for komplett remisjon (CR; 40-60%) enn azacitidin, men toksisiteten er høy, varighet av remisjonen kort, og det fins ikke evidens for å gi konsolideringskurer (Hast et al., 2003; Knipp et al., 2007; Vittayawacharin et al., 2023).
Lavdosert kjemoterapi (MDS)
Sist faglig oppdatert: 21.12.2023
Bakgrunn
Lavdosert kjemoterapi har ikke vist overlevelsesgevinst eller redusert AML-transformasjon i en uselektert gruppe pasienter. Unntaksvis kan lavdosert cytarabin anbefales til enkeltpasienter for å redusere leukocytose og/eller blastandel i benmargen og for å snu en cytopeniutvikling.
Melfalan kan ha effekt i en spesiell pasientgruppe som beskrives nedenfor.
Melfalan
5 studier med 98 inkluderte pasienter viste at pasienter med høy-risiko MDS eller AML med hypocellulær benmarg og fravær av ugunstig cytogenetikk, kan oppnå respons ved lavdosert melfalan. 51 pasienter i studiene responderte, 31 med komplett respons. CR varer ca. 12 måneder. Melfalan 2 mg tablett gis daglig i 8 uker; meget god toleranse og få bivirkninger (Denzlinger et al., 2000; Stratmann et al., 2019; Whittle et al., 2013).
En retrospektiv studie fra 2019 med 31 eldre pasienter med relapsert eller refraktær normo/hypocellulær AML, viste at lavdosert melfalan hadde effekt hos pasienter med intermediær ELN-klassifikasjon som tidligere hadde fått hypometylerende behandling.
Indikasjon: Symptomgivende hypo-/normocellulær høyrisiko MDS eller MDS-AML, med fravær av flere enn to cytogenetiske avvik eller kromosom 7 forandringer. Dersom hypometylerende behandling ikke er forsøkt, bør dette vurderes som et behandlingsalternativ før oppstart med melfalan tabletter.
Dosering: Melfalan 2 mg daglig fram til respons, maksimalt 8 uker. Ved residiv kan melfalan behandlingen startes på nytt (i samme dosering) og vil kunne gi ny komplett remisjon. Remisjonsvarigheten vil oftest vare kortere. Ved neste recidiv, kan det gjøres ytterlgiere forsøk, men remisjonsperioden pleier å bli kortere for hver gang. Nye cytogenetiske avvik kan utvikles i forløpet, særlig delesjon 17p med mutasjon i TP53.
Studier (MDS)
Sist faglig oppdatert: 21.12.2023
Alle pasienter med behandlingstrengende MDS som ikke er aktuelle for allo-HCT bør vurderes med tanke på inklusjon i studier.
Se hjemmesiden for Norsk hematologisk selskap med tanke på aktuelle studier eller sjekk ved universitetssykehusene om studier pågår.
Introduksjon KMML
Sist faglig oppdatert: 21.12.2023
Definisjon
KMML er en undergruppe av myelodysplastiske/myeloproliferative neoplasier (Khoury et al., 2022). KMML kjennetegnes av monocytose med varierende grad av myeloproliferative og myelodysplastiske trekk. Det er sentralt å utelukke andre årsaker til monocytose (infeksjoner, autoimmune sykdommer mm). Hos ca 25% av KMML pasientene sees ledsagende autoimmunitet/systemisk inflammasjon (Moreno Berggren et al., 2021; Zahid et al., 2017).
I WHO 2016 klassifikasjonen ble KMML definert ved persisterende monocytose i perifert blod ≥ 1 x109/L (>3 mnd) og monocyttene måtte utgjøre utgjøre ≥ 10 % av leukocyttene I WHO 2022 klassifikasjonen er den absolutte grensen for monocytter senket til ≥ 0,5 x109/L, mens det opprettholdes at monocyttene skal utgjøre ≥ 10 % av leukocyttene i perifert blod. Det er ikke lenger krav om persisterende monocytose > 3 mnd (Khoury et al., 2022). De diagnostiske kriteriene avviker noe mellom ICC og WHO 2022 (Arber et al., 2022). Se tabell 2.1.2 for klassifisering i henhold til WHO 2016, WHO 2022 og ICC 2022.
| WHO 2016 | WHO 2022 | ICC 2022 |
|---|---|---|---|
Hovedkriterier |
|
|
|
Bi kriterier |
|
|
|
Diagnose krav | De 4 første kriteriene må være til stede. Punkt 5 kan mangle hvis de 4 første og et av de følgende er tilstede:
| Alle hovedkriterier må være til stede.
| I tilfeller uten påvist klonalitet kreves monocytose (≥1 x 109/L) og
|
Forekomst og etiologi
KMML er en sjelden sykdom med insidens ca. 0.4/100 000/år (Pleyer et al., 2021). Den rammer primært eldre > 60 år, median alder ved diagnosetidspunkt er 73–75 år, og den er hyppigere hos menn enn hos kvinner (ratio rundt 2:1). 10 % av tilfellene er terapi-relatert (Patnaik et al., 2022).
Diagnostikk, utredning og klasifikkasjonssystemer (KMML)
Sist faglig oppdatert: 21.12.2023
Diagnostikk og utredning ved KMML
Symptomer og kliniske funn
MD-KMML og MP-KMML presenterer seg ofte forskjellig klinisk (Patnaik et al., 2022).
MD-KMML har hyppigere cytopenier, som kan medføre blødningstendens, gjentatte infeksjoner og behov for transfusjoner, mens MP-KMML har tegn på prolifirativ sykdom med forstørret lever og milt samt konstitusjonelle symptomer som feber, nattesvette, utmattelse og skjelettsmerter.
20- 25 % av KMML pasientene har tidligere, samtidige eller påfølgende autoimmune sykdommer eller udefinerte systemiske inflammatoriske tilstander som Sweet syndrom, erythema nodosum, vasculitt, muskel- og ledd smerter; se eget kapittel) (Zahid et al., 2017). Leukemiske hudinfiltrater og serøse effusjoner (pleurale, perikardielle og peritoneale) kan forekomme. I tillegg er KMML er den hyppigste assosierte neoplasi ved systemisk mastocytose (obs. maculopapuløst hudutslett; cutan mastocytose; gjerne kalt urticaria pigmentosa).
Blodprøver
- Hb, trombocytter, hvite med differensialtelling, blodutstryk, MCV, LD, haptoglobin, bilirubin, reticulocytter, ASAT, ALAT, ALP, kreatinin, CRP, immunglobuliner, serumelektroforese, ANA og lysozym (enzym som forekommer i monocytter, særlig i udifferensierte monocytter)
- Flowcytometri av perifert blod (heparinglass) kan påvise monocyttforandringer (CD14+, CD16-) i over 94% som er et diagnostisk kriterium ved diagnosetidspunktet i.h.t. WHO klassifikasjonen
- Differensialdiagnostiske prøver for å utelukke andre årsaker til monocytose (tuberkulose, sarkoidose, kroniske soppinfeksjoner, subakutte endokarditter, leishmaniasis, SLE o.l.)
- Lysozym (ved nyresvikt).
- Tryptase hvis assosiert systemisk mastocytose mistenkes (kan være forhøyet ved KMML)
Molekylærpatologiske blodprøver
- RT-PCR/FISH mot BCR-ABL1 (utelukke KML)
- JAK2, CALR og MPL (vil støtte diagnosen myeloproliferativ neoplasi framfor KMML)
- Mutert kit (D816V) (ved mistanke om systemisk mastocytose)
Benmargsundersøkelser
Benmargsaspirat til:
- Morfologisk vurdering
- Immunfenotyping/flowcytometri (heparinglass)
- Cytogenetisk undersøkelser (McCoy-glass)
- Molekylærpatologiske undersøkelse (EDTA-glass):
A. Bare hvis eosinofili: PDGFRA, PDGFRB, FGFR1 og PCM1-JAK2
B. NGS myeloid panel: Hos alle som er aktuell for allo-HCT og hos andre hvis det forventes å få behandlingsmessige konsekvenser. Bør tas før oppstart med cytostatika.
Morfologisk vurdering
Benmargsaspirat
- KMML karakteriseres av monocytose i blod og benmarg. Ofte fins både normale og abnorme monocytter
- Abnorme monocytter (ofte mer umodne): De er granulerte, har tettere kromatin og mer uttalte kjerneinnbuktninger, folder og mer grålig cytoplasma enn promonocytter og monoblaster
- Monoblastene er store og har rund kjerne. Kjernen har distinkte nukleoler. Cytoplasma kan inneholde vacuoler og fine granula
- Promonocyttene har mer irregulære og noe mer foldede kjerner
- Disse 3 typer monocyttoide celler (patologiske monocytter, promonocytter og blaster) med ulik modenhetsgrad kan være vanskelige å skille fra hverandre
- Dertil kan abnorme monocytter (med granula i cytoplasma) og promonocytter være vanskelige å skille fra dysplastiske hypogranulære myelocytter og promyelocytter
- Ved KMML telles promonocytter, monoblaster og myeloblaster som blaster
- Hvis blaster og promonocytter utgjør 20 %, foreligger AML
- Dysplasi i de myeloide rekkene er som beskrevet for MDS
Benmargsbiopsi
- Undersøkelsen er av betydning for vurdering av dysplastiske/ patologiske megakaryocytter, cellularitet, grad av fibrose og eventuelt funn av differensialdiagnoser eller oppdagelse av samtidig forekommende systemisk mastocytose.
Flowcytometri
- Andel klassiske monocytter i blod (CD14+, CD16-) over 94% er et diagnostisk kriterium i WHO 2022. Flowcytometri i blod bør derfor gjøres hos alle pasienter ved mistanke om KMML
- De flowcytometriske avvikene som kan sees i benmargen ved KMML er for en stor del de samme som beskrevet for MDS som fenotypiske avvik både i myeloide precursorceller, i monocytt- og granulocytt-linjen samt relativ andel CD34+ celler, promonocytter og monocytter.
- Avvik på monocyttene ved KMML kan som ved MDS være økt ekspresjon av CD56, avvikende ekspresjon av CD2 og ofte redusert ekspresjon av HLA-DR, CD64, CD36 m.m (Feng et al., 2018).
Genetiske analyser
Cytogenetiske avvik
Cytogenetiske avvik forekommer hos 20–30 %. Hyppigst forekommende er trisomi 8, -Y, kromosom 7 defekter (monosomi 7 og del(7q), trisomi 21 og kompleks karyotype.
NGS myeloid panel
Somatiske mutasjoner påvises hos over 90 %. TET2 mutationer forekommer i ca. 60 % av pasientene, SRSF2 i ca. 50 % ASXL1 i ca. 40 % og RAS ca 30 % (Patnaik et al., 2022).
Mutasjoner i ASXL1, RUNX1, NRAS and SETBP1 har prognostisk verdi og er inkludert i CPSS-mol risiko vurdering (Elena et al., 2016). ASXL1 mutasjon har i mange studier vist å være assosiert med dårlig prognose, mens TET2 mutasjon (en eller flere) i fravær av mutasjon i ASXL1 er assosiert med gunstig prognose (Itzykson et al., 2013). CMML-MP er hyppig assosiert med signalveien RAS med NRAS, KRAS, CBL) and JAK2V617F mutasjoner.
En liten andel (3-5%) av KMML pasienter har NPM1 mutasjon. Disse er karakterisert ved å ha svært rask progresjon til AML, kjemorefraktær sykdom og dårlig prognose.
Klassifisering av KMML
WHO 2022-/ICC 2022 klassifikasjonene skiller i likhet med WHO 2016 klassifikasjonen mellom en myelodysplastisk form (MD-KMML) med leukocytt tall < 13 x 109/L og en myeloproliferativ form (MP-KMML) med leukocytt tall ≥ 13 x 109/L. Denne inndelingen har klinisk, behandlingsmessig og prognostisk betydning. MP-KMML har dårligere prognose (Patnaik et al., 2022).
KMML-0 er fjernet i WHO 2022 og ICC 2022 klassifikasjonene siden den prognostiske betydningen anses å være liten. Det skilles nå mellom KMML-1 (<5% blaster i perifert blod og < 10 % blaster i benmarg) og KMML-2 (5-19% blaster i perifert blod og 10-19% i benmarg eller tilstedeværelse av Auer staver). Se tabell 12.15.2.
Pasienter med klonal monocytose som ikke oppfyller kriterie for KMML eller annen hematologisk tiltstand, klassifiseres som klonal monocytose av usikker signifikans (CMUS) Diagnostiske kriterier for CMUS er angitt i tabell 12.15.1. Pasienter med CMUS bør følges som MDS pasienter med CCUS.
Tabel 12.15.1 Diagnostiske kriterier for Klonal monocytose av usikker signifikans (CMUS):
- Persisterende monocytose ≥0.5 x 109/L som må utgjøre 10% eller mer av antall leukocytter.
- Ingen cytopeni tilstede.
- Mutasjon i minst ett gen assosiert med MDS med allelfrekvens (VAF) ≥2%.
- Ingen økning av blaster eller andre morfologiske funn forenlig med KMML i benmargen.
- Pasienten oppfylinger ikke kriterier for annen hematologisk neoplasi.
- Reaktiv monocytose er utelukket.
KMML klassifikasjon | Blastandel i blod* | Blastandel i benmarg* |
|---|---|---|
WHO 2016 |
|
|
KMML-0 | < 2 % | < 5 % |
KMML-1 | 2–4 % | 5–9 % |
KMML-2 | 5–19 % | 10–19% eller Auer staver |
|
|
|
WHO 2022/ICC 2022 |
|
|
KMML-1 | <5 % | <10 % |
KMML-2 | 5–19 % | 10–19% eller Auer staver |
Prognostiske scoringssystemer (KMML)
Sist faglig oppdatert: 21.12.2023
Prognostiske scoringssystemer
Det er utviklet flere prognostiske scoringssystemer for KMML. IPSS-M er ikke validert for KMML og skal derfor ikke benyttes. Vi har valgt å bruke CPSS og CPSS-mol.
Siden MD-KMML er svært lik MDS, kan det vurderes å supplere med IPSS-M for tilleggsinformasjon selvsagt med viten om at den ikke er validert for MD-KMML.
CPSS: inkluderer parametrene blastandel i perifert blod og benmarg, leukocytt-tall i blod, transfusjonsbehov og cytogenetiske avvik, og scores som angitt i tabell 2.1.4
CPSS definerer 4 risikogruppene lav, intermediær-1, intermediær-2 og høy som angitt i tabell 2.1.7 (Such et al., 2013).
Prognostisk variabel |
| Poeng |
|
|---|---|---|---|
| 0 | 1 | 2 |
Andel blaster | Blod <5% BM <10% | Blod 5 til 19% BM 10 til 19% |
|
Leukocytt tall | < 13 x 109/L | ≥ 13 x 109/L |
|
Cytogenetikk* | Lav | Intermediær | Høy |
Transfusjonsbehov** | Nei | Ja |
|
*Cytogenetisk risiko klassifisering:
Lav: Normal eller isolert tap av kromosom Y.
Intermediær: Andre abnormaliteter.
Høy: Trisomi 8, kompleks karyotype (≥ 3 abnormaliteter) eller kromosom 7 anomalier.
**Definert som minst 1 erytrocyttenhet hver 8. uke over en periode på 4 måneder.
CPSS-Mol benytter samme parametre som CPSS, men inkluderer også 4 mutasjoner: ASXL1, NRAS, RUNX1 og SETBP1 (Elena et al., 2016). Disse mutasjonene kombinert med cytogenetisk risikogruppe i CPSS utgjør variabelen genetisk risikogruppe (tabell 2.1.5), som igjen integreres i en total score sammen med variablene blastandel i benmarg, leukocytt tall og transfusjonsavhengighet, se tabell 2.1.6.
Modellen identifiserer risikogruppene lav, intermediær-1, intermediær-2 og høy (Elena et al., 2016).Forskjellen mellom median overlevelse og transformasjon til AML ved bruk av CPSS og CPSS-mol fremgår av tabell 2.1.7 og 2.1.8).
Det kan bemerkes at både CPSS og CPSS-Mol er basert på pasienter med median alder 72-73 år. Disse pasientene har gjerne tilleggssykdommer som påvirke overlevelse, og risikoestimatene må derfor tolkes med varsomhet for yngre pasienter.
Det henvises til CPSS-mol kalkulator: qxmd.com
| CPSS cytogenetisk risikogruppe* | ASXL1 | NRAS | RUNX1 | SETBP1 | |
|---|---|---|---|---|---|---|
Variabel score |
|
|
|
|
| |
0 | Lav | Umutert | Umutert | Umutert | Umutert | |
1 | Intermediær | Mutert | Mutert | – | Mutert | |
2 | Høy | – | – | Mutert | – | |
Genetisk risikogruppe | Score |
|
|
|
| |
Lav | 0 |
|
|
|
| |
Intermediær-1 | 1 |
|
|
|
| |
Intermediær-2 | 2 |
|
|
|
| |
Høy | ≥ 3 |
|
|
|
| |
*CPSS cytogenetisk risiko gruppe er definert i henhold til tabell 2.1.4.
| Genetisk risikogruppe* | Blastandel i BM | Leukocytt tall | Transfusjons-behov** |
|---|---|---|---|---|
Variabel score |
|
|
|
|
0 | Lav | < 5 % | < 13 x 109/L | Nei |
1 | Intermediær-1 | ≥ 5 % | ≥ 13 x 109/L | Ja |
2 | Intermediær-2 | – | – | – |
3 | Høy | – | – | – |
CPSS-Mol risikogruppe | Score |
|
|
|
Lav | 0 |
|
|
|
Intermediær-1 | 1 |
|
|
|
Intermediær-2 | 2–3 |
|
|
|
Høy | ≥ 4 |
|
|
|
*Genetisk risiko gruppe er definert i henhold til tabell 2.1.5
**Definert som minst 1 erytrocytt enhet hver 8. uke over en periode på 4 måneder.
Risikogruppe | Score | Overall survival (median) mnd | AML transformasjon (%) | |
|---|---|---|---|---|
2 år | 5 år | |||
Lav | 0 | 72 | 7 | 13 |
Intermediær-1 | 1 | 31 | 14 | 29 |
Intermediær-2 | 2–3 | 13 | 37 | 60 |
Høy | 4–5 | 5 | 73 | 73 |
Risikogruppe | Score | Overall survival (median) mnd | AML transformasjon (4 år) % |
|---|---|---|---|
Lav | 0 | Ikke nådd | 0 |
Intermediær-1 | 1 | 64 | 3 |
Intermediær-2 | 2–3 | 37 | 21 |
Høy | 4–5 | 18 | 48 |
Anbefaling for diagnose og prognose av pasienter med KMML:
- Alle pasienter bør klassifiseres i henhold til WHO 2022 og ICC 2022 klassifikasjonen
- Myeloide mutasjoner (NGS) må sjekkes hos alle pasienter som skal stamcelletransplanteres. Det bør også gjøres hos andre hvis det forventes å få behandlingsmessige konsekvenser
- Alle pasienter bør risikostratifiseres i henhold til CPSS, og alle transplantasjonsaktuelle pasienter også etter CPSS-mol
Behandling og oppfølging - sammendrag (KMML)
Sist faglig oppdatert: 21.12.2023
Vurder først om det er:
- behandlingsindikasjon
- aktuelt med allo-HCT
Behandlingsindikasjon:
- Feber, vekttap / avmagring, symptomatisk splenomegali, cytopeni, leukocytose og sykdomsprogresjon med økende blasttall.
- Andre leukemiske manifestasjoner, som gingival hyperplasi, leukemiske infiltrater i huden, lavgradig DIC eller alvorlig DIC-fibrinolyse og autoimmune fenomener
- Ulike forhold knyttet både til pasients helsetilstand som alder, funksjonsnivå, komorbiditet, kognitiv funksjon, livskvalitet og forventninger og også bør vurderes ved valg av behandling (Patnaik et al., 2022).
Avvente behandling/observasjon/oppfølgning hos:
Asymptomatiske pasienter med cytopenier uten blastøkning og uten vesentlig leukocytose. Det er ingen definerte cytopeninivåer for behandlingsstart, men ofte brukes samme kriterier som ved MDS: Hb <10 g/L, trombocytter < 30 x 109/L eller blødning.
Behandlingsalgoritme ved KMML:
- Vurder kurativ behandling med allo-HCT hvis akseptabel komorbiditet
Ingen øvre aldersgrense, men vurder nøye forventet effekt opp mot risikofaktorer - For transplantasjonsaktuell pasient, ikke vent med forberedende tiltak eller forbehandling
- Ønsket blasttall før allo-HCT < 5 %
Ikke-transplantasjonsaktuelle pasienter:
- Vurder om pasienten kan delta i studie
- MD-KMML: Vurder behandling med AZA
- MP-KMML: Hydroxyurea ev. kombinert AZA
- Lav-risk KMML og anemi: ESA (som ved MDS)
- Gi støttebehandling. Oppretthold akseptabel Hb. Tenk livskvalitet!
- Endelig behandlingsvalg er pasientens etter god informasjon
Allogen stamcelletransplantasjon ved KMML
Sist faglig oppdatert: 21.12.2023
Bakgrunn
Allo-HCT er den eneste kurative behandlingen ved KMML, men er pga. høy alder ved diagnose og ledsagende comorbiditet (pga. alder), kun aktuelt for en mindre pasientgruppe. Dertil er allo-HCT ved KMML forbundet med høy risiko for transplantasjonsrelaterte komplikasjoner eller recidiv. Det fins ingen prospektive studier. Responsraten i retrospektive studier strekker seg fra 17 til 50% med korresponderende transplantasjonsrelatert mortalitet (12-52%) og residivfrekvens (17-50%) (Patnaik et al., 2022). I en av de største retrospektive studiene med 513 KMML pasienter (median alder 53 år) fra EBMT var 4 års relapse free survival (RFS) 27%, og OS 33%. CR ved transplantasjonstidspunktet var assosiert med bedre RFS og OS. 64 nordiske (inkludert norske) pasienter med KMML er nylig samlet. Estimert overlevelse etter 5 år var 46.5% (35-61%), kumulativ transplantasjons-relatert mortalitet var 30% (19-42%) og kumulativt recidivfrekvens 25% (15-36%) etter 5 år (Wedge et al., 2021).
Vedrørende pasientenes komorbiditet og egnethet for allo-HCT, henvises til allo-HCT ved MDS. Siden resultatene etter allo-HCT ved KMML er dårligere enn ved MDS, må det vurderes nøye om pasienten totalt sett vil være tjent med allo-HCT. Transplantasjon i tidlig sykdomsfase kan ikke anbefales. I en retrospektiv studie kom pasienter med lav- og intermediær risk-1 (CPSS) dårligere ut enn ikke-transplanterte pasienter (Robin et al., 2022).
Pasienter med KMML aktuelle for allo-HCT
- Ta tidlig stilling til om det er indikasjon for all-HCT
- Vurder sykdommens prognose opp mot morbiditet
- Hvis pasienten er aktuell for allo-HCT: HLA-typ pasienten og start søk etter stamcellegiver.
Indikasjon for allo-HCT:
- KMML-2
- CPSS-mol høy og intermediær-2 risiko
- CPSS-mol intermediær-1 kan i enkelte tilfeller vurderes hos yngre pasienter.
Cytoreduksjon før allo-HCT:
Det fins ingen prospektive studier som kan bekrefte betydningen av å redusere blastandelen før allo-HCT. Bridging er fortsatt kontroversielt og anbefalinger er stort sett ektrapolert fra MDS der debulking til <10% blastandel før transplantasjon anvendes (omtales under MDS). KMML data fra EBMT registeret støtter indirekte betydningen av pretransplantasjons debulking siden eneste risikofaktor assosiert med overlevelse i denne serien var oppnådd CR før transplantasjon (Symeonidis et al., 2015). Det er ikke vist forskjell mellom hypomethylerende midler og intensiv kjemoterapi. Behandlingsvalg er avhengig av pasientens alder, comobiditet og cytogenetiske/molekylærgenetiske karakteristika. Dynamikken i sykdommen er viktig for behandlingsvalget. Det bør tilstrebes å gi så få behandlinger med kjemoterapi som mulig innen allo-HCT (Damaj et al., 2012; Gerds et al., 2012; Vittayawacharin et al., 2023).
Det må sendes søknad til Nasjonal gruppe for allo-HCT (OUS) for pasienter aktuelle for allo-HCT.
Azacitidin (AZA)
Ikke-proliferativ KMML (MD-KMML) er inkludert i mange MDS studier med HMA og har lignende respons som MDS. Det er nylig publisert en stor retrospektiv studie med KMML pasienter som viser overlevelsesgevinst for KMML med høyt blasttall (Pleyer et al., 2021). Azacitidin er kun godkjent for MDS-KMML, men denne studien inkluderte også MP-KMML med blastøkning. Således er AZA også er alternativ for pasienter med MP-KMML med høyt blasttall. AZA bør da ev. kombineres ved hydroxyurea.
Hydroxurea (HU)
En randomisert studie med HU vs. Etoposide viste overlegen respons ved HU (60 % vs. 36 %). Overlevelse i HU armen var 20 måneder sammenlignet med 9 måneder i etoposid armen. Responsene var imidlertid kortvarige.
HU anbefales som førstelinjebehandling for eldre pasienter med KMML-1 der hovedmålet er å redusere symptomene. Forlenget overlevelse kan ikke forventes. Det finnes ikke noe fastsatt nivå for leukocytt-tall eller miltstørrelse som kan anbefales som det optimale nivået for å introdusere behandling. Beslutningen bør baseres på pasientens symptomer og komorbiditet ADDIN EN.CITE.DATA ADDIN EN.CITE.DATA (Itzykson et al., 2018).
Bivirkninger ved HU er mildere enn med AZA. Hvis pasienten ikke responderer på HU eller utvikler sykdomsprogresjon, kan AZA vurderes.
Erytropoiestin (ESA)
Indikasjon anemi. Gis som ved MDS.
Autoimmune – og inflammatoriske sykdommer assosiert med MDS og KMML
Sist faglig oppdatert: 21.12.2023
10 – 25% pasienter med MDS/KMML (oftere ved KMML enn MDS) har assosiert systemisk inflammatorisk - eller autoimmun sykdom slik som polykondritt, polyartritt, thyreoidea sykdommer, nøytrofil dermatose, vaskulitt og bindevevssykdommer. Symptomer og tegn på dette kan inntre før og etter diagnosen med MDS / KMML (Hochman et al., 2022; Mekinian et al., 2016). Prognosen hos MDS/KMML pasienter med eller uten autoimmun sykdom varierer i ulike studier fra å være dårligere til forbedret (Hochman et al., 2022). I en meta-analyse fra 18 studier med 4603 pasienter fant Liang et al en kortere overlevelse for pasientene med inflammatorisk /autoimmun sykdom (Liang et al., 2022).
- Autoimmune cytopenier assosiert med MDS/KMML er uvanlig.
- Autoimmun hemolytisk anemi har blitt rapportert hos ca 3% av MDS pasienter og ses oftest ved lavrisiko MDS (Van Rhee et al., 1991).
- Immunologisk trombocytopeni ses også sjeldent ved MDS (Park et al., 2003).
Behandling av MDS/KMML-relatert autoimmun sykdom
Behandlingsindikasjon og type behandling må ta hensyn til subtype autoimmun sykdom, sykdommens alvorlighetsgrad, residiv frekvens og behov for spesifikk hematologisk behandling av underliggende MDS/KMML (Jachiet et al., 2021). Underliggende cytopenier og økt infeksjonsrisiko kan være utfordrende.
Behandlingsalternativer:
- Steroider. De fleste oppnår initialt god respons, men 50-70% får residiv eller steroid avhengighet (Fain et al., 2007).
- Azacytidin. Erfaringsmessig god effekt. En fransk studie viste komplett eller partiell autoimmun/autoinflammatorisk respons hos 87% av pasientene med MDS/KMML relatert autoimmun/autoinflammatorisk sykdom. Responsene kom etter en median på 3 sykluser (Jachiet et al., 2021).
- Ved manglende på sykdomsrettet behandling kan andre medikamenter med suppressive eller immunmodulerende medikamenter som metotrexate, azatioprim, mycophenolate mofetil eller cyklofosfamid), biologisk behandling (Rituximab, anti-TNF-alfa, tocilizumab, anakinra) (Jachiet et al., 2021). Effekten er dog usikker og hematologisk toksisitet utfordrende.
- Biologisk behandling kan ha dårligere effekt hos pasienter med MDS relatert autoimmun sykdom sammenliknet med pasienter med autoimmun sykdom som ikke er relatert til MDS (Mekinian et al., 2017).
- Vexas (Vacuoler. E1-enzym, X-bundet, Autosomatisk og Somatisk) syndrom er en autoinflammatorisk tilstand assosiert med feber, chondritt, tromboctyopeni og dysplastiske trekk i benmarg med vakuoler i benmargsceller. Tilstanden skyldes en ervervet mutasjon i UBA1 genet og fanges ikke opp av myeloid panel, men må bestilles separat. Tilstanden er responsiv til høydose steroider, men ingen DMARDs (disease modyfying antirheumatic drugs).
- Vexas syndrom behandles med JAK2 hemmer og/ eller allo-HCT. Hyppigst forekomst hos middelaldrende menn.
Medikamenter ikke registrert for MDS eller KMML
Sist faglig oppdatert: 21.12.2023
Decitabin
Decitabin kan vurderes ved intoleranse for azacitidin. I Norge er decitabin godkjent for pasienter med primær eller sekundær AML som ikke er kandidater for standard induksjons-behandling. Små retrospektive studier viser ingen klinisk signifikant effekt av å bytte til decitabin ved svikt på azacitidin (Lee et al., 2013). Noen studier tyder på at decitabin (5 eller 10 dagers kur) gir høyere responsrate hos pasienter med TP53-mutasjoner (Welch et al., 2016). Andre studier har ikke kunnet vise dette (Platzbecker, 2019). Pga. mangel på prospektive og randomiserte studier, anbefales ikke det ene hypometylerende medikamentet fremfor det andre ved mutert TP53.
Venetoklaks
Venetoklaks er en potent hemmer av bcl-2 (et antiapoptotisk protein) og er godkjent for AML i Norge. På bakgrunn av fase 2 studier som viser høy responsrate, er den godkjent i USA ved AML sammen med hypometylerende midler eller lavdose cytarabin. I flere av de publiserte AML-publikasjonene inngår pasienter med høy-risiko MDS og KMML (DiNardo et al., 2019; Wei et al., 2019).
Det er foreløpig kun presentert abstrakter eller små retrospektive singelsenterstudier av fase 1 og 2 studier for bruk av venetoklaks sammen med hypometylerende behandling ved MDS. De viser imidlertid høy responsrate og komplett remisjon hos ca 30 % (Azizi et al., 2020; Liu et al., 2020). Effekt inntrer tidligere enn ved hypometylerende behandling alene. Venetoklax har effekt både i 1. behandlingslinje, men også etter behandlingssvikt ved HMA, dvs. hos pasienter uten behandlingstilbud (Ball et al., 2020; Bazinet et al., 2022).
Venetoklaks har blitt brukt sammen med HMA som bro til allo-SCT (Bewersdorf et al., 2021; Jädersten et al., 2022; Yang et al., 2022). Fordelen fremfor AZA alene er en raskere og dypere effekt, samt mulighet for effekt ved AZA-resistens.
Studier taler for en høy risiko for benmargssuppresjon ved dosering som ved AML. Pågående studier for sammenligning av ulike doser og tidsvarighet pågår. Det kan være aktuelt å gi venetoklaks til enkelte pasienter ved residiv etter allo-HCT eller ved manglende respons på primærbehandling for om mulig å få pasienten i tilstrekkelig remisjon slik at allo-HCT kan utføres (Jädersten et al., 2022).
Dosering: Venetoklaks tabletter bør gis i kombinasjon med azacitidin eller decitabin. Doseringen bør være 400 mg daglig i 2 uker i 28 dagers sykluser. 200 mg daglig i 2 uker kan vurderes ved hypocelleulær benmarg. Tumor lyse forekommer sjelden slik at opptrapping av dosen ved oppstart som ved AML, anses unødvendig. Ved cytopeni, vurder benmargen etter en cyklus. Ved manglende effekt etter 2 sykluser med venetoklaks, er sannsynligheten for effekt vesentlig redusert. Avslutning bør vurderes spesielt ved alvorlig cytopeni.
Blodkreft i allmennpraksis
Generelle betraktninger
Sist faglig oppdatert: 23.12.2021
Antall nye tilfeller av blodkreft i Norge er lavt i forhold til de store kreftsykdommer som ca. mammae, ca prostatae, lungekreft og colorektal kreft. Allikevel er det en god del som lever med enten kurert blodkreft, eller med indolent sykdom som har stor risiko for progresjon eller tilbakefall.
Allmennpraktikeren ser noen av disse pasientene med jevne mellomrom. De trenger diagnostisering, oppfølging under behandling, kontroller og eventuelt palliativ behandling. Noen er unge ved diagnosetidspunktet og forventes å leve lenge.
Diagnostisering, valg av behandling og initial oppfølging etter ferdigbehandlet sykdom, er i stor grad sentralisert til universitetsykehus og større sykehus i landet. Detaljene rundt de enkelte behandlingsregimene er beskrevet i øvrige kapitler i dette handlingsprogrammet, men er for omfattende for daglig bruk i allmenpraksis.
Det er derfor her laget et eget kapittel som omhandler allmenlegens rolle i pasientforløpet til blodkreftpasienter med vekt på henvisningsrutiner, samhandling med sykehus under aktiv behandling og oppfølging av ferdig behandlede pasienter.
Primærhelsetjenestens viktigste rolle for denne pasientgruppen
Sist faglig oppdatert: 23.12.2021
- Henvise de rette pasientene til utredning ut fra anamnese, symptomer og funn. For akutt leukemi, kronisk lymfatisk leukemi og myelomatose er det innført pakkeforløp med definerte forløpstider som monitoreres. Se www.helsedirektoratet.no
- Støttesamtaler underveis i behandlingen (mange unge pasienter og unge familier).
- Primærlegens kunnskap om geografi, familierelasjoner, arbeidsmuligheter, organisering av hjemmetjenesten etc. er viktig med tanke på lokalt hjelpebehov og tilbud.
- Veiledning i sosiale og trygdemedisinske rettigheter ikke bare under aktiv sykdom, men også i rekonvalesensfasen. (se kapittel 14 Psykososiale forhold, hjelpetiltak og fysioterapi).
- Delta i overvåkning av hematologiske parametere under cytostatikabehandling.
- Oppfange og behandle bivirkninger under cellegift- og strålebehandling i aktiv behandlingsfase, særlig infeksjoner.
- Samarbeide med sykehus om kontrollene ved langvarig sykdom.
- Overta kontrollene etter at kontrollene ved sykehus er avsluttet. Ved slik etterkontroll er overvåking av symptomer med tanke på residiv sentralt.
- Diagnose og behandling av senskader (stråleskader, stoffskifte, parestesier, fatigue).
I tillegg må primærhelsetjenesten bistå med revaksinering der dette er anbefalt.
Utredning i allmennpraksis
Sist faglig oppdatert: 23.12.2021
Akutte leukemier debuterer ofte med symptomer på infeksjon, anemi og økt blødningstendens. Residiverende øvre luftveis- eller hudinfeksjoner, sårdannelse i munnslimhinne, feber av ukjent årsak, slapphet og vekttap er vanlige, men uspesifikke symptomer. Hvis slike symptomer er ledsaget av anemi sammen med leukopeni/leukocytose med blaster i blod og trombocytopeni, er det ofte grunn til rask (øyeblikkelig hjelp) videre utredning. Nyoppstått spontan blødningstendens er et alarmsymptom. Ved mistanke om akutt leukemi henvises til utredning i pakkeforløp.
Myelomatose debuterer oftest i almenpraksis med ryggsmerter. Samtidig har langt de fleste pasienter med ryggsmerter i almenpraksis andre, mer trivielle årsaker til smertene. Avtakende bruk av SR som screeningundersøkelse for malignitet i førstelinjetjenesten har kanskje ført til at myelomatosediagnosen noen ganger forsinkes i uheldig grad. Funn av anemi, hypercalcemi eller økt kreatinin bør skjerpe mistanken. Påvisning av m-komponent ved serumelektroforese og evt urinelektroforese kan være viktige prøver ved nyoppståtte ryggsmerter hos eldre. Ved mistanke om myelomatose henvises til utredning i pakkeforløp. Myelomatose skal behandles først når pasienten får symptomer. Stigende M-komponent alene skal ikke behandles, men bør henvises.
Hos noen eldre (5 % over 80 år) kan man finne en M-komponent i lav konsentrasjon ved serumelektroforese. Hvis M-komponenten er under 30 g/l og pasienten ikke har symptomer eller beinmargsvikt skyldes dette oftest monoklonal gammopati av usikker betydning (MGUS). Dette er en benign tilstand, men 1–2 %/år utvikler seg til myelomatose.
Hva bør fastlegen gjøre ved påvisning av en liten M-komponent?
Ved små M-komponenter (under 10 g/l), ingen symptomer, normal Hb, kreatinin, Ca, og urinelektroforese kan pasienten kontrolleres hos fastlege. Pasienten bør få beskjed om å kontakte legen ved symptomer (smerte, slapphet, nevrologiske symptomer). Kontroll hver 3. måned det første året, senere ved stabil tilstand ca en gang i året.
Ved høyere M-komponent eller usikkerhet bør pasienten henvises til hematolog/indremedisiner.
Kronisk lymfatisk leukemi og indolente lymfomer er ofte utbredt til mange lymfeknutestasjoner og/eller benmarg og vokser gjerne langsomt. Mange får stilt diagnosen pga. tilfeldig oppdagete avvik i hgb og leukocytter i blodprøver tatt i annen hensikt. Absolutt lymfocytose over 5 x 109/l med moden morfologi gir begrunnet mistanke om KLL.
Lymfeknutesvulsten er typisk uøm, fast/elastisk og forskyvelig mot underliggende vev og hud.
B-symptomer er sjeldnere enn ved aggressive lymfomer og Hodgkin.
- Nattesvette: gjentatt kraftig nattesvette siste måned.
- Feber: persisterende eller residiverende feber >38 siste måned uten kjent annen årsak.
- Vekttap: mer enn 10 % siste 6 måneder.
Ved siden av blodprøver til hgb, leukocytter, diff, trombocytter, CRP og LD er serum-elektroforese ofte en nyttig undersøkelse på evt. m-komponent ved mistanke om lymfom/KLL. Ved mistanke om KLL eller lymfom henvises til utredning i pakkeforløp.
Henvisningsrutiner til sykehus
Sist faglig oppdatert: 23.12.2021
Mikroskopisk og ofte også immunologisk og genetisk undersøkelse av blod, beinmarg og evt lymfeknutebiopsi er nødvendig for diagnose, klassifisering og valg av behandling av blodkreftsykdommer.
Ved mistanke om akutt leukemi skal pasienten henvises som øyeblikkelig hjelp, med telefonisk kontakt til aktuelle avdeling for blodsykdommer eller indremedisin til utredning i pakkeforløp
Ved mistanke om raskt voksende lymfeknutesvulst i mediastinum med ødem i ansikt/hals og evt besværet respirasjon må pasienten innlegges indremedisinsk avdeling som øyeblikkelig hjelp.
Billeddiagnostikk har relativt liten betydning initialt og kan forsinke forløpet.
Mistanke om kompresjon av medulla spinalis eller nerverøtter ved myelomatose, med distale pareser og sensibilitetstap, skal også føre til innleggelse som øyeblikkelig hjelp i sykehus som kan gi strålebehandling eller avlastende kirurgi.
Hvis allmennsymptomene dominerer det kliniske bildet, er det riktig å henvise til indremedisinsk avdeling/poliklinikk.
Henvisning til kirurgisk eller ØNH poliklinikk for biopsi eller cytologisk undersøkelse er viktigere enn radiologiske utredninger initialt ved klinisk mistenkelige lymfeknuter med mer fredelig klinikk.
Ved siden av opplysninger om kliniske funn og resultatet av relevante blodprøver (se over), er ofte opplysninger om tidligere verdier (hgb, hvite, trombocytter) av interesse for å kunne bedømme hastighet av sykdomsutviklingen.
Barn under 18 år henvises barneavdeling. Her må en bruke skjønn og tilpasse lokal praksis.
Se også kapittelet om forløpstider.
Oppfølging av blodkreft i allmennpraksis (kliniske tips)
Sist faglig oppdatert: 23.12.2021
Under aktiv behandling
Almenlegen skal ha epikrise med behandlingsplan. Oftest vil primærbehandlingen og kontrollen av denne foregå ved sykehus.
Hvis epikrise med plan for aktuell behandling fra sykehus inkluderer oppgaver for primærlegen, kan det være greit å gå gjennom epikrisen med pasienten og sikre at innholdet er forstått. Det bør legges en plan for kontroller / hematologisk overvåkning hos allmennlegen. Hvis avstanden til behandlende sykehus er stor, vil dette kunne hindre bomturer til sykehus ved interkurrent sykdom eller hvis blodverdiene er for lave til ny kur.
NB! Ta alltid kontakt med behandlende avdeling før kur utsettes eller behandlingsplan fravikes!
Infeksjonsovervåking og påvisning av neutropen feber er viktig. CRP-økning skyldes oftest bakteriell infeksjon, og ikke aktivitet i blodkreftsykdommen.
Leukocyttallet er typisk lavest 7–14 dager etter cytostatikakur. Pasienter med feber over 38 grader kombinert med leukocytter under 1 eller neutrofile under 0,5 skal som hovedregel innlegges og behandles som sepsis. Neutropeni uten feber og infeksjonssymptomer kan som oftest observeres, med temperaturmåling rektalt morgen og kveld. I palliativ fase vil det noen ganger være riktig å behandle infeksjoner i hjemmet med perorale antibiotika, selv om neutrofile er lave.
Pasienter med akutt leukemi og transplanterte pasienter har ofte tunnelert sentralt venekateter (CVK, Hickman), som brukes til blodprøvetaking, transfusjoner og intravenøs medikamentell behandling. Åpning og skylling av slike katetere er forbundet med risiko for luftemboli og infeksjon, og må bare gjøres av helsepersonell som har trening i dette, og følge fastlagte prosedyrer. Hvis slikt personell, prosedyrer og utstyr ikke er tilgjengelig, må man ta blodprøver og gi intravenøs behandling i perifer vene på armen ved behov i almenpraksis, selv om pasienten har innlagt CVK.
Transfusjoner administreres som hovedregel på sykehus. Kliniske symptomer er oftest avgjørende for transfusjonsindikasjonen. Ved hgb under 8 og cytostatikaindusert trombocytopeni under 10 bør transfusjon drøftes med sykehuset.
Allmennlege må kunne justere smertebehandling og kvalmebehandling. Se Nasjonalt handlingsprogram med retningslinjer for palliasjon i kreftomsorgen.
Pasienter som står på antikoagulasjonsbehandling får økt blødningsrisiko ved trombocytopeni som følge av behandling og sykdom. Ved trombocytter <50 bør evt. hematolog konsulteres for dosereduksjon. Det er ofte aktuelt å gå over fra peroral antikoagulasjon til (doseredusert) lavmolekylært heparin, og seponere i perioder med betydelig trombocytopeni.
Etter avsluttet behandling i sykehus
Revaksinasjon: Etter allogen stamcelletransplantasjon og høydosebehandling med autolog stamcellestøtte (HMAS) ved lymfom og myelomatose anbefales noen pasienter full revaksinering etter 6 mnd (stivkrampe, difteri, polio og kikhoste). Pneumokokk vaksine startesetter 3 mnd og seinere ca. hvert 5. år, avhengig av målbart serumantistofftiter. Levendeog perorale vaksiner bør unngåes i 2 år etter HMAS. Vaksinering bør initieres av ansvarlig sykehusavdeling.
Influensavaksinering kan gis etter skjønn og de generelle anbefalinger fra Folkehelsa.
Fatigue er en tilstand alle allmennleger bør kjenne til spesielt i relasjon til kreftpasienter, som får stråleterapi eller cytostatika. Tilstanden karakteriseres ved en subjektiv følelse av økt trettbarhet og nedsatt funksjonskapasitet, som ikke forsvinner ved hvile eller søvn. Fatigue beskrives av mange kreftpasienter som den mest belastende behandlingsrelaterte plagen, og er vanlig forekommende.
Vi har ikke gode data på hva som er beste behandling. Et lett treningsprogram ser ut til å være viktig for å bryte den onde sirkelen av symptomer som ofte er årsak til at ferdigbehandlede, friskmeldte kreftpasienter likevel ikke kommer seg tilbake i full jobb.
Overføring av kontrollene til primærhelsetjenesten
Etter behandling med helbredelse som mål av blodkreftsykdom er det noen ganger aktuelt å overføre kontrollene helt eller delvis til primærhelsetjenesten. Sykehusavdelingen som hadde ansvaret for primærbehandlingen bør legge en plan for slike kontroller. Planen bør inkludere informasjon om hvilke prøver og undersøkelser som skal tas, og om kontrollhyppighet.
Etter allogen stamcelletransplantasjon er overvåking med tanke på residiv av grunnsykdom, diagnostikk og behandling av kronisk transplantat-mot-vert sykdom og senskader sentralt.
Nyttige adresser / referanser for allmennpraktikere
Sist faglig oppdatert: 23.12.2021
Nasjonalt handlingsprogram med retningslinjer for palliasjon i kreftomsorgen.
Nasjonalt handlingsprogram for diagnostisering, behandling og oppfølging av maligne lymfomer
www.kreftforeningen.no
Her ligger mye pasientrettet informasjon. Kreftforeningen har også mange gode brosjyrer for ulike kreftformer som kan bestilles. Dette gjelder også hefter om trygderettigheter, «Håndbok for foreldre med kreftsyke barn», «Når foreldre dør», «Mor eller far har kreft» osv.
www.legemiddelhandboka.no
I kapitlet om kreftsykdommer finnes kortfattede beskrivelser av legemiddelbehandling av blodkreftsykdommer.
Generelle betraktninger
Sist faglig oppdatert: 23.12.2021
Antall nye tilfeller av blodkreft i Norge er lavt i forhold til de store kreftsykdommer som ca. mammae, ca prostatae, lungekreft og colorektal kreft. Allikevel er det en god del som lever med enten kurert blodkreft, eller med indolent sykdom som har stor risiko for progresjon eller tilbakefall.
Allmennpraktikeren ser noen av disse pasientene med jevne mellomrom. De trenger diagnostisering, oppfølging under behandling, kontroller og eventuelt palliativ behandling. Noen er unge ved diagnosetidspunktet og forventes å leve lenge.
Diagnostisering, valg av behandling og initial oppfølging etter ferdigbehandlet sykdom, er i stor grad sentralisert til universitetsykehus og større sykehus i landet. Detaljene rundt de enkelte behandlingsregimene er beskrevet i øvrige kapitler i dette handlingsprogrammet, men er for omfattende for daglig bruk i allmenpraksis.
Det er derfor her laget et eget kapittel som omhandler allmenlegens rolle i pasientforløpet til blodkreftpasienter med vekt på henvisningsrutiner, samhandling med sykehus under aktiv behandling og oppfølging av ferdig behandlede pasienter.
Primærhelsetjenestens viktigste rolle for denne pasientgruppen
Sist faglig oppdatert: 23.12.2021
- Henvise de rette pasientene til utredning ut fra anamnese, symptomer og funn. For akutt leukemi, kronisk lymfatisk leukemi og myelomatose er det innført pakkeforløp med definerte forløpstider som monitoreres. Se www.helsedirektoratet.no
- Støttesamtaler underveis i behandlingen (mange unge pasienter og unge familier).
- Primærlegens kunnskap om geografi, familierelasjoner, arbeidsmuligheter, organisering av hjemmetjenesten etc. er viktig med tanke på lokalt hjelpebehov og tilbud.
- Veiledning i sosiale og trygdemedisinske rettigheter ikke bare under aktiv sykdom, men også i rekonvalesensfasen. (se kapittel 14 Psykososiale forhold, hjelpetiltak og fysioterapi).
- Delta i overvåkning av hematologiske parametere under cytostatikabehandling.
- Oppfange og behandle bivirkninger under cellegift- og strålebehandling i aktiv behandlingsfase, særlig infeksjoner.
- Samarbeide med sykehus om kontrollene ved langvarig sykdom.
- Overta kontrollene etter at kontrollene ved sykehus er avsluttet. Ved slik etterkontroll er overvåking av symptomer med tanke på residiv sentralt.
- Diagnose og behandling av senskader (stråleskader, stoffskifte, parestesier, fatigue).
I tillegg må primærhelsetjenesten bistå med revaksinering der dette er anbefalt.
Utredning i allmennpraksis
Sist faglig oppdatert: 23.12.2021
Akutte leukemier debuterer ofte med symptomer på infeksjon, anemi og økt blødningstendens. Residiverende øvre luftveis- eller hudinfeksjoner, sårdannelse i munnslimhinne, feber av ukjent årsak, slapphet og vekttap er vanlige, men uspesifikke symptomer. Hvis slike symptomer er ledsaget av anemi sammen med leukopeni/leukocytose med blaster i blod og trombocytopeni, er det ofte grunn til rask (øyeblikkelig hjelp) videre utredning. Nyoppstått spontan blødningstendens er et alarmsymptom. Ved mistanke om akutt leukemi henvises til utredning i pakkeforløp.
Myelomatose debuterer oftest i almenpraksis med ryggsmerter. Samtidig har langt de fleste pasienter med ryggsmerter i almenpraksis andre, mer trivielle årsaker til smertene. Avtakende bruk av SR som screeningundersøkelse for malignitet i førstelinjetjenesten har kanskje ført til at myelomatosediagnosen noen ganger forsinkes i uheldig grad. Funn av anemi, hypercalcemi eller økt kreatinin bør skjerpe mistanken. Påvisning av m-komponent ved serumelektroforese og evt urinelektroforese kan være viktige prøver ved nyoppståtte ryggsmerter hos eldre. Ved mistanke om myelomatose henvises til utredning i pakkeforløp. Myelomatose skal behandles først når pasienten får symptomer. Stigende M-komponent alene skal ikke behandles, men bør henvises.
Hos noen eldre (5 % over 80 år) kan man finne en M-komponent i lav konsentrasjon ved serumelektroforese. Hvis M-komponenten er under 30 g/l og pasienten ikke har symptomer eller beinmargsvikt skyldes dette oftest monoklonal gammopati av usikker betydning (MGUS). Dette er en benign tilstand, men 1–2 %/år utvikler seg til myelomatose.
Hva bør fastlegen gjøre ved påvisning av en liten M-komponent?
Ved små M-komponenter (under 10 g/l), ingen symptomer, normal Hb, kreatinin, Ca, og urinelektroforese kan pasienten kontrolleres hos fastlege. Pasienten bør få beskjed om å kontakte legen ved symptomer (smerte, slapphet, nevrologiske symptomer). Kontroll hver 3. måned det første året, senere ved stabil tilstand ca en gang i året.
Ved høyere M-komponent eller usikkerhet bør pasienten henvises til hematolog/indremedisiner.
Kronisk lymfatisk leukemi og indolente lymfomer er ofte utbredt til mange lymfeknutestasjoner og/eller benmarg og vokser gjerne langsomt. Mange får stilt diagnosen pga. tilfeldig oppdagete avvik i hgb og leukocytter i blodprøver tatt i annen hensikt. Absolutt lymfocytose over 5 x 109/l med moden morfologi gir begrunnet mistanke om KLL.
Lymfeknutesvulsten er typisk uøm, fast/elastisk og forskyvelig mot underliggende vev og hud.
B-symptomer er sjeldnere enn ved aggressive lymfomer og Hodgkin.
- Nattesvette: gjentatt kraftig nattesvette siste måned.
- Feber: persisterende eller residiverende feber >38 siste måned uten kjent annen årsak.
- Vekttap: mer enn 10 % siste 6 måneder.
Ved siden av blodprøver til hgb, leukocytter, diff, trombocytter, CRP og LD er serum-elektroforese ofte en nyttig undersøkelse på evt. m-komponent ved mistanke om lymfom/KLL. Ved mistanke om KLL eller lymfom henvises til utredning i pakkeforløp.
Henvisningsrutiner til sykehus
Sist faglig oppdatert: 23.12.2021
Mikroskopisk og ofte også immunologisk og genetisk undersøkelse av blod, beinmarg og evt lymfeknutebiopsi er nødvendig for diagnose, klassifisering og valg av behandling av blodkreftsykdommer.
Ved mistanke om akutt leukemi skal pasienten henvises som øyeblikkelig hjelp, med telefonisk kontakt til aktuelle avdeling for blodsykdommer eller indremedisin til utredning i pakkeforløp
Ved mistanke om raskt voksende lymfeknutesvulst i mediastinum med ødem i ansikt/hals og evt besværet respirasjon må pasienten innlegges indremedisinsk avdeling som øyeblikkelig hjelp.
Billeddiagnostikk har relativt liten betydning initialt og kan forsinke forløpet.
Mistanke om kompresjon av medulla spinalis eller nerverøtter ved myelomatose, med distale pareser og sensibilitetstap, skal også føre til innleggelse som øyeblikkelig hjelp i sykehus som kan gi strålebehandling eller avlastende kirurgi.
Hvis allmennsymptomene dominerer det kliniske bildet, er det riktig å henvise til indremedisinsk avdeling/poliklinikk.
Henvisning til kirurgisk eller ØNH poliklinikk for biopsi eller cytologisk undersøkelse er viktigere enn radiologiske utredninger initialt ved klinisk mistenkelige lymfeknuter med mer fredelig klinikk.
Ved siden av opplysninger om kliniske funn og resultatet av relevante blodprøver (se over), er ofte opplysninger om tidligere verdier (hgb, hvite, trombocytter) av interesse for å kunne bedømme hastighet av sykdomsutviklingen.
Barn under 18 år henvises barneavdeling. Her må en bruke skjønn og tilpasse lokal praksis.
Se også kapittelet om forløpstider.
Oppfølging av blodkreft i allmennpraksis (kliniske tips)
Sist faglig oppdatert: 23.12.2021
Under aktiv behandling
Almenlegen skal ha epikrise med behandlingsplan. Oftest vil primærbehandlingen og kontrollen av denne foregå ved sykehus.
Hvis epikrise med plan for aktuell behandling fra sykehus inkluderer oppgaver for primærlegen, kan det være greit å gå gjennom epikrisen med pasienten og sikre at innholdet er forstått. Det bør legges en plan for kontroller / hematologisk overvåkning hos allmennlegen. Hvis avstanden til behandlende sykehus er stor, vil dette kunne hindre bomturer til sykehus ved interkurrent sykdom eller hvis blodverdiene er for lave til ny kur.
NB! Ta alltid kontakt med behandlende avdeling før kur utsettes eller behandlingsplan fravikes!
Infeksjonsovervåking og påvisning av neutropen feber er viktig. CRP-økning skyldes oftest bakteriell infeksjon, og ikke aktivitet i blodkreftsykdommen.
Leukocyttallet er typisk lavest 7–14 dager etter cytostatikakur. Pasienter med feber over 38 grader kombinert med leukocytter under 1 eller neutrofile under 0,5 skal som hovedregel innlegges og behandles som sepsis. Neutropeni uten feber og infeksjonssymptomer kan som oftest observeres, med temperaturmåling rektalt morgen og kveld. I palliativ fase vil det noen ganger være riktig å behandle infeksjoner i hjemmet med perorale antibiotika, selv om neutrofile er lave.
Pasienter med akutt leukemi og transplanterte pasienter har ofte tunnelert sentralt venekateter (CVK, Hickman), som brukes til blodprøvetaking, transfusjoner og intravenøs medikamentell behandling. Åpning og skylling av slike katetere er forbundet med risiko for luftemboli og infeksjon, og må bare gjøres av helsepersonell som har trening i dette, og følge fastlagte prosedyrer. Hvis slikt personell, prosedyrer og utstyr ikke er tilgjengelig, må man ta blodprøver og gi intravenøs behandling i perifer vene på armen ved behov i almenpraksis, selv om pasienten har innlagt CVK.
Transfusjoner administreres som hovedregel på sykehus. Kliniske symptomer er oftest avgjørende for transfusjonsindikasjonen. Ved hgb under 8 og cytostatikaindusert trombocytopeni under 10 bør transfusjon drøftes med sykehuset.
Allmennlege må kunne justere smertebehandling og kvalmebehandling. Se Nasjonalt handlingsprogram med retningslinjer for palliasjon i kreftomsorgen.
Pasienter som står på antikoagulasjonsbehandling får økt blødningsrisiko ved trombocytopeni som følge av behandling og sykdom. Ved trombocytter <50 bør evt. hematolog konsulteres for dosereduksjon. Det er ofte aktuelt å gå over fra peroral antikoagulasjon til (doseredusert) lavmolekylært heparin, og seponere i perioder med betydelig trombocytopeni.
Etter avsluttet behandling i sykehus
Revaksinasjon: Etter allogen stamcelletransplantasjon og høydosebehandling med autolog stamcellestøtte (HMAS) ved lymfom og myelomatose anbefales noen pasienter full revaksinering etter 6 mnd (stivkrampe, difteri, polio og kikhoste). Pneumokokk vaksine startesetter 3 mnd og seinere ca. hvert 5. år, avhengig av målbart serumantistofftiter. Levendeog perorale vaksiner bør unngåes i 2 år etter HMAS. Vaksinering bør initieres av ansvarlig sykehusavdeling.
Influensavaksinering kan gis etter skjønn og de generelle anbefalinger fra Folkehelsa.
Fatigue er en tilstand alle allmennleger bør kjenne til spesielt i relasjon til kreftpasienter, som får stråleterapi eller cytostatika. Tilstanden karakteriseres ved en subjektiv følelse av økt trettbarhet og nedsatt funksjonskapasitet, som ikke forsvinner ved hvile eller søvn. Fatigue beskrives av mange kreftpasienter som den mest belastende behandlingsrelaterte plagen, og er vanlig forekommende.
Vi har ikke gode data på hva som er beste behandling. Et lett treningsprogram ser ut til å være viktig for å bryte den onde sirkelen av symptomer som ofte er årsak til at ferdigbehandlede, friskmeldte kreftpasienter likevel ikke kommer seg tilbake i full jobb.
Overføring av kontrollene til primærhelsetjenesten
Etter behandling med helbredelse som mål av blodkreftsykdom er det noen ganger aktuelt å overføre kontrollene helt eller delvis til primærhelsetjenesten. Sykehusavdelingen som hadde ansvaret for primærbehandlingen bør legge en plan for slike kontroller. Planen bør inkludere informasjon om hvilke prøver og undersøkelser som skal tas, og om kontrollhyppighet.
Etter allogen stamcelletransplantasjon er overvåking med tanke på residiv av grunnsykdom, diagnostikk og behandling av kronisk transplantat-mot-vert sykdom og senskader sentralt.
Nyttige adresser / referanser for allmennpraktikere
Sist faglig oppdatert: 23.12.2021
Nasjonalt handlingsprogram med retningslinjer for palliasjon i kreftomsorgen.
Nasjonalt handlingsprogram for diagnostisering, behandling og oppfølging av maligne lymfomer
www.kreftforeningen.no
Her ligger mye pasientrettet informasjon. Kreftforeningen har også mange gode brosjyrer for ulike kreftformer som kan bestilles. Dette gjelder også hefter om trygderettigheter, «Håndbok for foreldre med kreftsyke barn», «Når foreldre dør», «Mor eller far har kreft» osv.
www.legemiddelhandboka.no
I kapitlet om kreftsykdommer finnes kortfattede beskrivelser av legemiddelbehandling av blodkreftsykdommer.
Metode og prosess
Bakgrunn
Sist faglig oppdatert: 21.12.2023
Mange onkologiske faggrupper la i en årrekke ned et betydelig arbeid for å komme frem til konsensusbaserte faglige anbefalinger for diagnostikk og behandling av ulike typer kreft. Som ledd i Nasjonal strategi for kreftområdet (2006–2009) fikk Helsedirektoratet i oppdrag å videreutvikle og oppdatere faggruppenes anbefalinger til nasjonale handlingsprogrammer for kreftbehandling, i nært samarbeid med fagmiljøene, de regionale helseforetakene, Nasjonalt kunnskapssenter for helsetjenesten, og andre relevante myndigheter. De nasjonale handlingsprogrammene representerer en videreføring og en formalisering av faggruppenes anbefalinger.
Helsedirektoratet rettet i januar 2009 en henvendelse til Norsk Selskap for Hematologi og ba om forslag til representanter til en arbeidsgruppe. Alle nødvendige faggrupper var representert og gruppen besto av fagfolk fra alle helseregioner. Første utgave av Nasjonalt handlingsprogram med retningslinje for diagnostikk, behandling og oppfølging av maligne blodsykdommer ble utgitt i samarbeid med Helsedirektoratet i 2012. Dette er den tiende oppdateringen av handlingsprogrammet. Se tabell nederst for deltagere. Helsedirektoratet takker arbeidsgruppene for stor innsats i oppdateringen av handlingsprogrammet.
Formål med anbefalingene
Sist faglig oppdatert: 21.12.2023
Nasjonale handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft (Nasjonale handlingsprogram) skal bidra til at det offentlige tilbudet i kreftomsorgen blir av god kvalitet og likeverdig over hele landet.
Rettslig betydning
Sist faglig oppdatert: 21.12.2023
Helsedirektoratet skal utvikle, formidle og vedlikeholde normerende produkter, det vil si nasjonale faglige retningslinjer, nasjonale veiledere, nasjonal faglige råd og pakkeforløp, som understøtter målene for helse- og omsorgstjenesten.
Nasjonale anbefalinger og råd skal baseres på kunnskap om god praksis, og skal bidra til kontinuerlig forbedring av virksomhet og tjenester, jf. spesialisthelsetjenesteloven § 7-3, helse- og omsorgstjenesteloven § 12-5 og folkehelseloven § 24.
Nasjonale anbefalinger og råd setter en norm for hva som er faglig forsvarlig. Anbefalinger/råd utgitt av Helsedirektoratet er ikke rettslig bindende, men er faglig normerende for valg man anser fremmer kvalitet, god praksis og likhet i tjenesten på utgivelsestidspunktet.
I situasjoner der helsepersonell velger løsninger som i vesentlig grad avviker fra gitte anbefalinger, skal dette dokumenteres, jf. journalforskriften § 6, bokstav g. Helsepersonell bør være forberedt på å begrunne sine valg i eventuelle klagesaker eller ved tilsyn.
Målgrupper
Sist faglig oppdatert: 21.12.2023
Målgrupper for de nasjonale handlingsprogrammene er spesialister innen medisin, kirurgi, onkologi, radiologi og patologi. De vil også være av interesse for allmennleger, pasienter, pårørende og andre aktører på kreftfeltet.
Omfang og avgrensning
Sist faglig oppdatert: 21.12.2023
Dette handlingsprogrammet omtaler diagnostikk og behandling for følgende sykdomsgrupper:
- Akutt myelogen leukemi
- Akutt lymfoblastisk leukemi/lymfoblastisk lymfom
- Myelodysplastisk syndrom
- Kronisk myelogen leukemi
- Myeloproliferative sykdommer
- Myelomatose
- Amyloidose
- Kronisk lymfatisk leukemi
- Morbus Waldenstrøm og andre indolente lymfomer.
- Følgende kreftformer er ikke omhandlet i dette handlingsprogrammet:
- Lymfekreft, som i Norge for det meste behandles av spesialister i kreftsykdommer (onkologi)
Se: Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne lymfomer - Blodkreft hos barn
Se Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn - Det har ikke vært brukerrepresentanter med i arbeidsgruppene.
Handlingsprogram og Nye metoder
Sist faglig oppdatert: 21.12.2023
De regionale helseforetakene har ansvar for Nye metoder, et felles system for å beslutte hvilke metoder som kan tilbys i spesialisthelsetjenesten. Nye metoder er et prioriteringsverktøy. Beslutningene i Nye metoder gjelder nasjonalt, les mer her: Bakgrunn: Hvorfor har vi Nye metoder?
Handlingsprogram og andre normerende produkter kan ikke anbefale legemidler eller andre metoder som ikke har finansering i den offentlige helsetjenesten. Handlingsprogrammene oppdateres i overenstemmelse med beslutninger i Nye metoder.
Kunnskapsbasert metode
Sist faglig oppdatert: 21.12.2023
Kunnskapsbasert praksis innebærer at både forskningsbasert kunnskap, erfaringsbasert klinisk kunnskap og pasientenes/ brukernes ønsker og behov vurderes samlet. I utarbeidelsen og revidering av nasjonale handlingsprogram har det ikke vært en tradisjon for brukermedvirkning.
Innholdet i en retningslinje skal baseres på et oppdatert, gjennomarbeidet og dokumentert kunnskapsgrunnlag. Det innebærer at forskningslitteratur, klinisk erfaring og brukererfaring vurderes opp mot ønskede og uønskede konsekvenser av et tiltak. På områder der det er funnet mindre forskningsbasert kunnskap, og/eller overføringsverdien fra internasjonal til norsk helsetjeneste er lav, blir klinisk kunnskap og brukerkunnskap tillagt større vekt.
Dette nasjonale handlingsprogrammet inneholder svært mange anbefalinger. Det er ikke praktisk mulig, innenfor gitte ressursrammer, å samle all relevant forskningsbasert kunnskap for hver anbefaling. Den forskningsbaserte kunnskapen er hovedsakelig basert på forskning identifisert i anerkjente internasjonale retningslinjer og nyere studier. Det er heller ikke praktisk mulig å gjennomgående bruke GRADE eller SIGN-metodikken for gradering av kunnskapsgrunnlaget.
Handlingsprogram og andre normerende produkter skal også forholde seg til prioriteringskriteriene og ressursbruk. Med ressurser menes direkte kostnader, men også personell og tidsbruk.
Grad av normering
Sist faglig oppdatert: 21.12.2023
Grad av normering angis i teksten i hver enkelt anbefaling/råd:
- Når Helsedirektoratet skriver «skal», betyr det at anbefalingen/rådet er basert på lov eller forskrift, eller når det er så klart faglig forankret at det sjelden er forsvarlig å ikke gjøre som anbefalt.
- Når det står bør» eller «anbefaler», er det en sterk anbefaling/råd som vil gjelde i de aller fleste tilfeller.
- Når det står «kan» eller «foreslår» er det en svak anbefaling/råd der ulike valg kan være riktig for ulike pasienter.
Relaterte normerende produkter
Sist faglig oppdatert: 21.12.2023
Arbeidsform og deltagere
Sist faglig oppdatert: 21.12.2023
Handlingsprogrammet oppdateres av en gruppe oppnevnt av Helsedirektoratet. Representantene i gruppen er utnevnt av de regionale helseforetakene. Etter Helsedirektoratets bearbeidelse og gjennomgang av utkast til handlingsprogram, blir handlingsprogrammet publisert på Helsedirektoratets temaside om kreft
Handlingsprogrammet ble publisert desember 2023 i revidert utgave.
Leder for utarbeiding av handlingsprogrammet overlege Tor Henrik Andersen Tvedt, OUS.
Tabell over hovedforfattere og medforfattere - oppdateringsgruppe 2023:
Leder for handlingsprogramgruppen: Tor Henrik Andersen Tvedt, Oslo Universitetssykehus Rikshospitalet
Kapittel | Kapittelleder | Medforfattere |
|---|---|---|
Diagnostikk | Ikke oppdatert nå | Signe Spetalen Lars Helgeland |
AML | Tor Henrik Andersen Tvedt (Kun ett nytt legemiddel tatt inn) | Andrea Lenartova Anne Sophie von Krogh Eivind A. Myhre
|
ALL | Petter Quist Paulsen, St. Olavs hospital | Hilde S. Wik Anders Vik Anette Eilertsen |
Myeloproliferative sykdommer | Hoa Tran | Diaa Hassaf Håkon Reikvam Eivind Galteland |
KLL | Geir Tjønnfjord (Kun ett nytt legemiddel tatt inn) | Emadoldin Feyzi |
Myelomatose | Fredrik Schjesvold | Galina Tsykunova Nina Guldbrandsen Einar Haukås Jürgen Rolke Øyvind Hjertner Tobias Schmidt Slørdahl |
Amyloidose | Galina Tsykunova (Små tekstlige endringer) | Tor Henrik Anderson Tvedt Fredrik Schjesvold Nina Guldbrandsen Øyvind Hjertner |
KML | Henrik Hjort-Hansen | Tobias Gedde-Dahl Waleed Ghanima |
| Andre indolente lymfoprofilerative sykdommer | Ikke oppdatert nå Sigbjørn Berentsen | Geir E. Tjønnfjord |
| Myelodysplastike syndromer (MDS) og kronisk myelo-monocytt-leukemi (KMML) | Ingunn Dybedal | Maryan M.Ali Astrid Bergrem Birgitte Eiken Hilde Jensvoll Hedda Lerdal Heidi Lona Emil Nyquist Liv Osnes Marit Rinde Kristoffer Sand Signe Spetalen Robert Brudevold Synne Torkildsen Camilla Vo |
| Blodkreft i allmennpraksis | Ikke oppdatert nå
| |
Psykososiale forhold, hjelpetiltak og fysioterapi | Ikke oppdatert nå |
Habilitet
Sist faglig oppdatert: 21.12.2023
Hovedforfattere for de reviderte kapitlene har fylt ut Helsedirektoratets habilitetsskjema. Ingen interesser med konsekvenser for deltakelse i arbeidet er identifisert. Skjemaene er arkivert i Helsedirektoratets arkiv, og innsyn kan gis ved forespørsel.
Kontaktpersoner i Helsedirektoratet
Sist faglig oppdatert: 21.12.2023
Kontaktperson i Helsedirektoratet er seniorrådgiver Hege Wang, avdeling spesialisthelsetjenester
Fagansvarlig på kreftområdet er spesialrådgiver Sissi Espetvedt: sissi.leyell.espetvedt@helsedir.no