






Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av sarkom
Forord
Sist faglig oppdatert: 28.02.2022
Nasjonale handlingsprogrammer for kreftbehandling skal bidra til at det offentlige tilbudet i kreftomsorgen blir av god kvalitet og likeverdig over hele landet. Målgrupper for retningslinjene er leger og legespesialister innen medisin, kirurgi, onkologi, radiologi og patologi. De vil også være av interesse for allmennleger, pasienter og pårørende. Handlingsprogrammet vil etter hvert suppleres med veiledning for allmennpraktikere, sykepleiere og andre faggruppers arbeid med kreftpasienter. Målet er å dekke hele pasientforløpet.
Nasjonale retningslinjer fra Helsedirektoratet er å betrakte som anbefalinger og råd, basert på oppdatert faglig kunnskap som er fremskaffet på en systematisk, kunnskapsbasert måte. De nasjonale retningslinjene gir uttrykk for hva som anses som god praksis på utgivelsestidspunktet, og er ment som et hjelpemiddel ved de avveininger tjenesteyterne må gjøre for å oppnå forsvarlighet og god kvalitet i tjenesten. Nasjonale retningslinjer er ikke direkte rettslig bindende for mottagerne, men bør langt på vei være styrende for de valg som skal tas. Ved å følge oppdaterte nasjonale retningslinjer vil fagpersonell bidra til å oppfylle kravet om faglig forsvarlighet. Dersom en velger løsninger som i vesentlig grad avviker fra de nasjonale retningslinjene, bør en dokumentere dette og være forberedt på å begrunne sine valg. Sykehusenes eiere og ledelse bør tilrettelegge virksomheten slik at de nasjonale retningslinjene kan følges.
Innholdet i de nasjonale retningslinjene for sarkom vil vurderes årlig, og om nødvendig oppdateres.
Helsedirektoratet takker arbeidsgruppen for stor innsats i utarbeidelsen av de nasjonale retningslinjene. Vi håper retningslinjene vil være et nyttig arbeidsredskap for spesialister som behandler pasienter med sarkom.
Nasjonalt handlingsprogram med retninglinjer for diagnostikk, behandling og oppfølging av sarkom ble publisert første gang 14. juni 2012.
Denne utgaven av Nasjonalt handlingsprogram for sarkom er den fjerde utgaven, publisert 28. februar 2022.
Bjørn Guldvog
Helsedirektør
Innledning
Sist faglig oppdatert: 28.02.2022
Sarkomer er maligne svulster som oppstår i bindevev og utgjør omtrent 1 % av alle nye krefttilfeller. Svulstene oppstår oftest i ekstremiteter, trunkus, abdominalt og retroperitonealt. Det diagnostiseres omtrent 50 nye bensarkomer og 500 bløtvevssarkomer hvert år i Norge. Pasienter i alle aldersgrupper kan bli rammet av sarkom, men hos barn og tenåringer er sarkom den nest hyppigste formen for kreft i solid vev utenfor sentralnervesystemet.
Optimal behandling krever tverrfaglig samarbeid mellom kirurger, radiologer, nukleærmedisinere, patologer, onkologer og andre spesialister. Behandlingen varierer fra kun kirurgi til noe av den mest intensive behandlingen som gis kreftpasienter, med ulike kombinasjoner av kirurgi, strålebehandling og cellegift. Pasientgruppen er liten og heterogen, og det er et utstrakt nasjonalt og internasjonalt samarbeid om behandlingsanbefalinger og kliniske studier. De norske behandlingssentrene har et bredt internasjonalt samarbeid, der det skandinaviske samarbeidet i Skandinavisk sarkomgruppe (SSG; www.ssg-org.net) er sentralt. Europeiske grupper og europeiske retningslinjer er også viktige grunnlag for utformingen av norske retningslinjer ADDIN EN.CITE.DATA ADDIN EN.CITE.DATA (Casali et al., 2022; Gronchi et al., 2021; Strauss et al., 2021).
Lindrende behandling er ikke vesensforskjellig fra andre krefttyper. Det erderfor ikke gitt detaljerte retningslinjer for palliativ behandling i dette handlingsprogrammet, og det vises til Nasjonalt handlingsprogram for palliasjon i kreftomsorgen (Nasjonalt handlingsprogram for palliasjon i kreftomsorgen: nasjonal faglig retningslinje, 2019).
Organisering av sarkomomsorgen
Sist faglig oppdatert: 28.02.2022
Universitetssykehusene i Oslo, Bergen, Trondheim og Tromsø har etablert hvert sitt sarkomsenter som utreder og behandler de fleste pasientene i sin region. Kirurgi ved bensarkom er sentralisert til Bergen og Oslo ved Flerregional behandlingstjeneste for kirurgi ved bensarkom.
Multidisiplinært team (MDT)
Alle sarkomsentrene har etablert et eget multidisiplinært team (MDT) for sarkom.Det utnevnes en klinisk leder som har det faglige behandlingsansvaret, og dette ansvaret bør nedfelles i klinikerens arbeidsinstruks. Lederen bør være et aktivt medlem av kjerneteamet.
Et sarkom MDT bør ha et kjerneteam bestående av:
- Kirurger/ortopeder og onkologer som har sin kliniske virksomhet hovedsakelig innen sarkombehandling.
- Radiologer, nukleærmedisinere og patologer med spisskompetanse i sarkomutredning.
- Pediatrisk onkolog med erfaring innen behandling av svulster i ben og bløtvev.
Hvert MDT bør etablere fast samarbeid med andre kliniske spesialiteter (thorakskirurger, nevrokirurger, ØNH kirurger, kontaktpersoner for partikkelbestråling) som tilkalles til MDT-møter ved behov. Det bør være minst 2–3 leger med spesialkompetanse innen hvert fagområde.
Et sarkom MDT bør ha en formell organisering, og den daglige virksomheten og ekstern/intern service bør ivaretas av en fast koordinator. Sarkom MDT bør ha regelmessige møter til faste tider, minst ukentlig. Alle pasienter med sarkom eller mesenkymale svulster med usikkert malignitetspotensiale der multidisiplinær behandling kan være aktuelt, eller diagnosen er usikker, bør diskuteres på sarkom MDT. Beslutning om tiltak bør baseres på anbefalinger fra MDT, og i samråd med pasienten. MDT-anbefalingene og endelig beslutning skal dokumenteres i journal, og det bør fremgå tydelig fra journalnotat at behandlingsplan er basert på MDT-anbefalingene.
Pasientene bør hospitaliseres i faste enheter der personell og støttepersonell har erfaring og kapasitet til å håndtere denne pasientgruppens spesielle utfordringer.
Norsk sarkomgruppe
Norsk sarkomgruppe er et tverrfaglig forum for fagpersoner som jobber med sarkompasienter. Gruppen består av ortopeder, gastrokirurger, gynekologer, onkologer, radiologer, nukleærmedisinere, patologer, fysioterapeuter, sykepleiere og assosierte medlemmer med forskjellige spesialiteter i alle helseregioner som deltar i utredning, behandling og rehabilitering av sarkompasienter.
Flerregional behandlingstjeneste for kirurgi ved bensarkom
Kirurgi ved bensarkom er sentralisert til Haukeland universitetssykehus og Oslo universitetssykehus ved Flerregional behandlingstjeneste for kirurgi ved bensarkom. De leverer årlige rapporteringer via RHFene til Helse- og omsorgsdepartementet.
Nasjonal kompetansetjeneste for sarkomer
Oslo universitetssykehus, Radiumhospitalet, har ansvar for Nasjonal kompetansetjeneste for sarkom som gir landsdekkende veiledning, informasjon og forskningsstøtte. Kompetansetjenesten leverer årsrapporter via Helse Sør-Øst RHF til Helse- og omsorgsdepartementet.
Pasientforening for sarkomer
Sist faglig oppdatert: 28.02.2022
Pasient- og støtteforeningen Sarkomer ble stiftet 1. oktober 2011, fordi det var et behov for å ha en forening som kunne tale sarkomrammedes sak. Foreningen er også en støtteforening som skal hevde pårørendes og etterlattes interesser. Hensikten med foreningen er å bedre sarkomrammedes velferd og øke oppmerksomheten om sarkom.
Foreningen driver med alt fra informasjonsvirksomhet om sarkom, deltakelse i høringer, brukermedvirkning og likepersonsarbeid. Likepersoner i Sarkomer er tidligere pasienter, pårørende og etterlatte som gir tips og råd til sarkomrammede. Pasientforeningen samarbeider med Sarkomforum for sykepleie og fysioterapi, OUS, og Nasjonal kompetansetjeneste for sarkomer.
Sarkomer sitter med en brukerrepresentant i referansegruppen for Nasjonal kompetansetjeneste for sarkom og Flerregional behandlingstjeneste for kirurgi ved bensarkom. Sarkomer ved daglig leder og styre er brukermedvirkere i en variert portefølje av forskning på sarkom, både medisinsk forskning og sosialmedisinsk forskning.
Sarkomer gir ut «Sarkomen» som er et populærvitenskaplig forum og medlemsnytt for medlemmene og andre interesserte. «Sarkomen» utkommer med 4 utgaver per år.
Et av de viktigste områdene for foreningen er å bidra til god informasjonsflyt om sarkom, slik at fastleger raskere kan henvise ved mistanke om sarkom. Sarkomer støtter kurs for sarkomopplæring av helsepersonell.
Sarkomer har utviklet og publisert en app med informasjon for sarkompasienter om diagnostikk, behandling, rehabilitering og oppfølging. Appen heter «Sarkom» og kan lastes ned gratis.
Foreningen har egen nettside: www.sarkomer.no, egen facebookside og Instagramkonto. Sarkomer ble assosiert medlem av Kreftforeningen 25. april 2013.
Epidemiologi
Sist faglig oppdatert: 28.02.2022
Sarkom er en sjelden krefttype og utgjør drøyt 1 % av alle maligne svulster. For oppdaterte tall på forekomst av sarkom i Norge henvises til årsrapporter fra Kvalitetsregister for sarkom, som publiseres av Kreftregisteret. For 2020 ble det rapportert 555 nye sarkomtilfeller i Norge, hvorav 53 var bensarkomer.
I og med at enkelte av undergruppene av sarkom er små kan det forekomme til dels store naturlige variasjoner i antall nye tilfeller fra år til år. Sarkom er relativt sett hyppigere hos barn og ungdom, og er den nest vanligste formen for malign solid svulst utenfor sentralnervesystemet i barnealder.
Bløtvevssarkom
Sist faglig oppdatert: 28.02.2022
Bløtvevssarkom opptrer i alle aldersgrupper. Gjennomsnittsalder er cirka 60 år. Sykdommen forekommer noe hyppigere hos menn enn kvinner. Bløtvevssarkom kan oppstå hvor som helst i kroppen, men de vanligste lokalisasjonene er fordøyelseskanalen og underekstremitetene.
Bløtvevsarkom i ekstremitet og trunkus
Bløtvevssarkom i ekstremitet og trunkus består av mange undertyper med en del fellestrekk, men med forskjellig biologisk oppførsel. De typene som forekommer hyppigst (C. Trovik et al., 2017) er:
- udifferensiert pleomorft sarkom (UPS)
- liposarkom
- leiomyosarkom
- myksofibrosarkom
- synovialt sarkom
Andre typer som forekommer sjeldnere er bl.a. rhabdomyosarkom, angiosarkom, malign perifer nerveskjedetumor (MPNST) og solitær fibrøs tumor.
Overlevelsen av pasienter med bløtvevssarkomer er relatert til histologisk diagnose, malignitetsgrad, metastaser ved diagnose, tumorstørrelse og lokalisasjon. 5-års metastasefri overlevelse for pasienter med lokalisert sykdom er rapportert å være omkring 70 % (C. Trovik et al., 2017). Pasienter som har påvist spredning ved diagnosetidspunkt har dårligere prognose.
Abdominalt og retroperitonealt sarkom
Bløtvevssarkom i abdomen og bekken utgjør 25–35 % av alle bløtvevssarkomer. De deles ofte inn etter lokalisasjon; retroperitonealt og intraabdominalt.
En retroperitoneal tumor har sitt utgangspunkt på bakre bukvegg, dorsalt for parietale blad av peritoneum, mellom diafragma og bekkenbunnen. Lyskekanalen og skrotum er i denne forbindelse å betrakte som en utløper av retroperitonealrommet og er et predileksjonssted for liposarkom. Retroperitoneale sarkomer utgjør cirka 15 % av det totale antall bløtvevssarkomer (P. W. T. Pisters, 2002). Etter radikal kirurgi er risikoen for lokalt (abdominalt) residiv større enn for fjernmetastaser, 30-60 % vs. 20 % (Gronchi et al., 2009; Lewis, Leung, Woodruff, & Brennan, 1998). Mange pasienter vil dø av lokalt residiv uten andre metastaser. Liposarkom og leiomyosarkom dominerer bildet, mindre hyppig er udifferensiert pleomorft sarkom (UPS), solitær fibrøs tumor (SFT) og malign perifer nerveskjedetumor (MPNST) (Brennan, Antonescu, Moraco, & Singer, 2014). En rekke andre sjeldne typer forekommer. Hvorvidt et bløtvevssarkom retroperitonealt har andre egenskaper enn svulster med tilsvarende histologi i ekstremiteter, er uavklart, og behandlingen er til dels forskjellig.
Intraabdominale sarkomer utgjøres hovedsakelig av gastrointestinal stromal tumor (GIST). Det ble i 2020 rapportert 146 nye tilfeller av GIST i Norge. GIST kan oppstå hvor som helst i mage-tarmkanalen, fra spiserør til anus. Omkring 60 % oppstår i magesekken og omkring 30 % i tynntarm. I sjeldne tilfeller oppstår de utenfor mage-tarmkanalen, f.eks. i omentet og retroperitonealt. GIST oppstår som regel sporadisk, men kan i sjeldne tilfeller være knyttet opp til syndromer, som Carneys triade og nevrofibromatose type 1.
Gynekologisk sarkom
Gynekologisk sarkom rammer kvinner i alle aldre. Sykdommen utgjør omkring 5 % av alle kreftsykdommer i uterus. Gynekologiske sarkomer forekommer som regel i livmoren, og den klart vanligste histologiske undertypen er leiomyosarkom, fulgt av endometriestromasarkom og adenosarkom. Karsinosarkom var tidligere klassifisert som sarkom, men er nå klassifisert som et høygradig malignt endometriekarsinom og omtales derfor ikke her.
Sarkom i mamma
Sarkom i mamma forekommer sjelden og utgjør mindre enn 1 % av all brystkreft og under 5 % av alle bløtvevssarkomer. Malign phyllodestumor og angiosarkom er de største undergruppene.
Phyllodestumor er en sarkomatoid lesjon bestående av epiteliale komponenter og bindevevselementer som ved fibroadenomer, men med større celletetthet i den stromale komponenten. Phyllodestumor utgjør et bredt spekter av lesjoner og klassifiseres som benign, borderline eller malign. En benign phyllodestumor kan morfologisk være vanskelig å skille fra et fibroadenom. Phyllodestumor har en utpreget lokal residivtendens, og behandlingen for alle typer er komplett kirurgisk eksisjon med mikroskopisk frie marginer. Malign phyllodes utredes, behandles og følges opp som andre sarkomer i mamma.
Angiosarkomer i mamma kan være primære eller sekundære (radioterapi, lymfødem). Risikoen for stråleassosiert angiosarkom etter behandling for brystkreft er under 1 %, og høyeste insidens sees etter 5–10 år. Primært angiosarkom utgår fra endoteliale strukturer i kjertelvevet, mens sekundært angiosarkom oppstår i huden der tumor infiltrerer diffust, og kan være multifokal. Klinisk kan tilstanden forveksles med eksem og postirradiære forandringer. Sekundært angiosarkom er oftere høygradig enn primært angiosarkom.
Se også Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av pasienter med brystkreft (Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av pasienter med brystkreft: nasjonal faglig retningslinje, 2021).
Bensarkom
Sist faglig oppdatert: 28.02.2022
Samlet forekomst av bensarkom er cirka 50 nye tilfeller i Norge per år. Sarkom i ben består av mange ulike varianter. Hyppigst forekommer:
- Osteosarkom – malign tumor med bendifferensiering
- Ewing sarkom: småcellet, rundcellet malign tumor med differensiering i retning av nevroektodermalt vev
- Kondrosarkom – malign tumor med bruskdifferensiering
Andre sjeldne bensarkom er kordom, udifferensiert pleomorft sarkom, angiosarkom, leiomyosarkom, malign kjempecelletumor og adamantinom.
Det er omkring 15-20 nye tilfeller av osteosarkom hvert år. Sykdommen er vanligst hos unge i alderen 10–30 år, og gjennomsnittsalder ved diagnose er cirka 16 år. Osteosarkom forekommer noe oftere hos gutter enn hos jenter (Berner et al., 2015), og er oftest lokalisert til metafysen av lange rørknokler med hovedvekt rundt kne og skulder.
Kondrosarkom utgjør omkring 20 nye tilfeller årlig. Median alder på diagnosetidspunktet er 50 år. Sykdommen forekommer noe hyppigere hos menn enn hos kvinner. Kondrosarkomer er ofte lokalisert i bekkenet eller lårben nær hofteleddet, men forekommer også i ryggsøyle og overarmsben.
Ewing sarkom utgjør cirka 5–10 nye tilfeller hvert år. Median alder er 14 år, og sykdommen forekommer svært sjelden >40 år. Det er omtrent lik fordeling mellom kjønnene. Primærtumor er oftest lokalisert i diafysen av lange rørknokler, men forekommer også i det aksiale skjelettet (bekken og ryggsøyle).
Overlevelsen av pasienter med bensarkom er relatert til histologisk diagnose, metastatisk status ved diagnose, tumorstørrelse og lokalisasjon. 5-års overlevelse for pasienter med ikke-metastatisk osteogent sarkom og Ewing sarkom er ca 70 % (Zaikova et al., 2015). Prognosen for pasienter som har metastaser på diagnosetidspunkt er dårligere.
Tilstander som gir økt risiko for sarkom
Sist faglig oppdatert: 28.02.2022
De fleste sarkomer oppstår spontant uten kjent årsak, men enkelte medfødte tilstander og arvelige genfeil kan gi økt risiko for sarkomutvikling.
Retinoblastom
Pasienter med hereditært retinoblastom har en økt risiko for å utvikle sarkom senere i livet, spesielt osteosarkom (Eng et al., 1993; Kinge, Tranheim, & Eide, 2004; L. M. Smith & Donaldson). Familiært retinoblastom er forårsaket av en mutasjon i kjønnscellene i RB1 tumorsuppressorgenet.
Li-Fraumeni syndrom
Li-Fraumeni er et autosomalt dominant syndrom. Dette er forårsaket av mutasjon i suppressorgenet TP53. Affiserte personer har økt insidens av ulike typer svulster som brystkreft, osteosarkom, bløtvevssarkom, hjernesvulster, binyrebarkkarsinom og leukemi (Andreassen et al., 1993; J. Hung & Anderson, 1997; F. P. Li & Fraumeni, 1969; Malkin et al., 1992; Thoresen, 1992; Toguchida et al., 1992; Travis et al., 2006; Tsuchiya et al., 2000). Debutalder er lavere enn ved sporadisk kreft.
Nevrofibromatose
Opp til halvparten av maligne perifere nerveskjedesvulster sees hos pasienter med nevrofibromatose type 1 (NF1) (Goldblum, Weiss, & Folpe, 2019; Zhou et al., 2003). NF1 er en autosomal dominant tilstand som karakteriseres av mutasjon i NF1 genet, som er et tumorsuppressorgen lokalisert på kromosom 17. NF1 medfører også økt risiko for andre sarkomer, spesielt GIST.
Benigne benlidelser
Enkelte benlidelser som enkondromatose (Olliers sykdom og Mafuccis syndrom), multiple osteokondromer og Pagets sykdom gir økt risiko for bensarkom (Haibach, Farrell, & Dittrich, 1985; Huvos, Butler, & Bretsky, 1983; J. Liu, Hudkins, Swee, & Unni, 1987; Schwartz et al., 1987; Wick, Siegal, Unni, McLeod, & Greditzer, 1981).
Stråleassosiert sarkom
Sist faglig oppdatert: 28.02.2022
Stråleassosiert sarkom er en sjelden, men alvorlig komplikasjon etter strålebehandling (Brenner, Curtis, Hall, & Ron, 2000; Murray, Werner, Greeff, & Taylor, 1999; Yap et al., 2002). De kriterier som oftest benyttes for definisjon av stråleassosiert sarkom er (Arlen et al., 1971; Cahan, Woodard, Higinbotham, Stewart, & Coley, 1998; Murray et al., 1999):
- Strålebehandling skal være gitt og sarkomet skal ha oppstått i strålefeltet.
- Det skal ikke foreligge holdepunkter for at sarkomet var til stede før strålebehandling.
- Sarkomet skal være diagnostisert etter en latenstid på minst to år.
- Sarkomet skal morfologisk være bekreftet og være av en annen histologisk type enn primærtumor.
Visse svulster, slik som retinoblastom, er assosiert med en relativt høy frekvens av sekundærsvulster, uavhengig av om det er gitt strålebehandling eller ikke, men de stråleassosierte oppstår gjerne etter en kortere latenstid (Murray et al., 1999; L. M. Smith & Donaldson).
Stråleassosierte sarkomer utgjør 2,5–5,5 % av sarkomtilfeller (Brenner et al., 2000; Hawkins et al., 1996; Menu-Branthomme et al., 2004; Murray et al., 1999). Latenstiden etter strålebehandling er median ca. 14 år (fra 2–60 år) (Bjerkehagen et al., 2008). De hyppigste histologiske diagnoser er udifferensiert pleomorft sarkom (UPS), osteosarkom, angiosarkom og malign perifer nerveskjedetumor. Det er uvanlig å se liposarkom og sarkomer med patognomoniske kromosomtranslokasjoner eller mutasjoner, for eksempel småcellet rundcellet sarkom, synovialt sarkom eller gastrointestinal stromal tumor (GIST). Prognosen for stråleassosierte sarkomer er ofte dårlig, med median rapportert overlevelsestid fra 12 til 48 måneder etter diagnose (Blanchard, Reynolds, Grant, Farley, & Donohue, 2002; Cha, Antonescu, Quan, Maru, & Brennan, 2004; Huvos, Sundaresan, Bretsky, & Butler, 1985; Lagrange et al., 2000; Murray et al., 1999; Souba et al., 1986), men hvis radikal kirurgi kan utføres initialt har pasienten lik prognose som ved sporadiske sarkom. Negative prognostiske faktorer for overlevelse er i tillegg metastaser ved diagnosetidspunkt, påviste tumornekroser, lokalisasjon sentralt og stor tumorstørrelse (Bjerkehagen et al., 2008).
Fastlegens rolle ved sarkom
Fastlegens rolle i diagnostikk og henvisning til pakkeforløp
Sist faglig oppdatert: 28.02.2022
Fastlegen vil ofte være den første av helsepersonell som møter pasienter med symptomer fra et sarkom. Det kan ofte være tidlig i sykdomsutviklingen, i en fase hvor symptomene er vage og uspesifikke. Å sette symptomer, varighet og funn inn i rett sammenheng er sentralt i den diagnostiske prosessen. Det er laget egne diagnoseveiledere for klargjøring av inklusjonskriteriene i Pakkeforløp for kreft, se Diagnoseveileder sarkom - Helsedirektoratet (Sarkom)
Bensarkom bør mistenkes ved palpabel tumor i knokkel eller ved uklar diagnose og vedvarende dype murrende smerter og nattesmerter uten annen forklaring.
Bløtvevssarkom i ekstremiteter, trunkus eller ØNH-regionen skal mistenkes ved:
- Alle dype svulster uansett størrelse, immobile mot kontrahert muskulatur.
- Alle subkutane svulster over 5 cm.
- Alle malignitetssuspekte svulster som vokser, tilbakefall etter tidligere kirurgi uansett histologi, uvanlige smerter eller andre symptomer.
Retroperitoneale og abdominale sarkomer gir ofte ingen eller kun diffuse plager og er som regel store når de blir oppdaget. Symptomene er ofte vanskelige å skille fra andre abdominale tilstander som smerter, anemi og abdominal oppfylling.
Beslutning om begrunnet mistanke om sarkom og henvisning til pakkeforløp baseres på samlet vurdering av symptomer, objektive kliniske funn og resultater av MR, eventuelt røntgen eller CT-undersøkelse.
Begrunnet mistanke om sarkom i ben og bløtvev i ekstremiteter, trunkus eller øre/nese/hals- region oppstår ved:
- Suspekt funn på røntgen av skjelett.
- Malignitetssuspekte forandringer i ben eller bløtvev ved MR.
- Uventet histologisk diagnose sarkom i kirurgisk preparat, biopsi eller cytologi (FNAC).
- Klinisk mistanke om tilbakefall hos tidligere sarkompasienter.
Begrunnet mistanke om retroperitoneale eller abdominalt sarkom oppstår ved:
- Funn av retroperitoneal tumor ved CT- eller MR-undersøkelse uten annen primærtumor.
- Funn av tumor i viscera hvor sarkom mistenkes radiologisk.
- Uventet funn av sarkom ved histologisk undersøkelse av fjernet organ.
Ved begrunnet mistanke henvises pasienten til pakkeforløp for sarkom. Det skal fremgå tydelig av henvisningen hva som utløser begrunnet mistanke om sarkom, samt eventuelt biopsisvar.
Fastlegens rolle for pasienter med sarkom i aktiv behandling
Sist faglig oppdatert: 28.02.2022
Fastlegens rolle i denne fasen vil variere, alt etter pasientens kliniske situasjon, livssituasjon og avstand til sykehus. Fastlegen bør være tilgjengelig for å bistå i oppfølging av behandlingen der det er hensiktsmessig for pasienten.
Innholdet kan være
- Smerter og komplikasjoner etter kirurgi.
- Hematologisk overvåkning mellom eventuelle cytostatikakurer.
- Håndtere bivirkninger av cytostatisk behandling.
- Justere smerte og kvalmebehandling.
- Gi råd om ernæring.
- Håndtere bivirkninger av strålebehandling.
- Avdekke eventuell tromboembolisk sykdom.
- Følge opp sykmelding og utskrivning av resepter.
Fastlegen kan bidra i behandling av angst, depresjon og andre psykiske plager, samt søvnvansker. Fastlegen kan også bidra til ivaretagelse av berørte familiemedlemmer, herunder eventuelle barn, og samarbeide med hjemmetjenesten der dette er aktuelt. Eventuell sykmelding eller forskriving av medikamenter bør skje i regi av den pårørendes fastlege.
Fastlegens rolle for pasienter som er i kontrollopplegg etter kurativ behandling for sarkom
Sist faglig oppdatert: 28.02.2022
Handlingsprogrammet anbefaler at de aller fleste sarkompasienter følges av spesialisthelsetjenesten etter fullført behandling. I enkelte tilfeller vil man likevel ut fra individuelle hensyn overføre kontrollansvaret til fastlegen, eksempelvis ved høy alder og lang reise til nærmeste sarkomsenter. Overføring og oppfølging hos fastlege må trygges gjennom tydelig informasjon og veiledning til pasient og fastlege. Overføringen må ivareta at fastlegene ikke har innkallingsrutiner og pasientens ansvar derfor blir større.
Utover de systematiske kontrollene som er beskrevet i kapittel 10, vil fastlegen fortsatt følge opp disse pasientene som ledd i vanlig klinisk praksis, og enkelte problemstillinger vil være knyttet til kreftsykdommen også utover rutinemessig oppfølging, for eksempel:
- Følge opp seneffekter etter kirurgi, strålebehandling og medikamentell behandling.
- Fange opp symptomer på tilbakefall og/eller spredning og henvise tilbake til ny utredning og behandling.
Fastlegens rolle for pasienter som er i rehabilitering
Sist faglig oppdatert: 28.02.2022
I alle deler av utredning, behandling og videre liv må fastlegen bistå i rehabiliteringen med forståelse og kunnskap om det som er viktig for pasienten. En sentral del av dette er å bistå med eventuell sykmelding og friskmeldingsprosess, og i samarbeid med sarkomsenter håndtere balansegangen mellom aktivitet og arbeidsliv på den ene siden og bivirkninger og seneffekter av behandlingen på den andre siden. Det vil i denne forbindelse også være aktuelt å henvise til spesialisert rehabilitering.
Fastlegens rolle ved tilbakefall og spredning av sarkom
Sist faglig oppdatert: 28.02.2022
Fastlegen må være oppmerksom på symptomer og tegn som kan indikere tilbakefall og spredning av sarkom. Symptomene kan være mange og ulike avhengig av anatomisk lokalisasjon. Det bør være lav terskel for bildediagnostikk ved nyoppståtte symptomer hos pasienter som tidligere er behandlet for sarkom.
Fastlegens håndtering tilpasses pasientens situasjon i kreftforløpet: For pasienter som er under oppfølging på sykehus, kontaktes avdelingen der oppfølgingen foregår. Dersom pasienten har avsluttet oppfølging på sykehus, henvises til rask ny utredning og behandling på sykehus.
Fastlegens rolle i arbeid med palliasjon og omsorg ved livets slutt
Sist faglig oppdatert: 28.02.2022
Flertallet av pasienter med utbredte metastaser fra sarkom er i en ikke-kurabel situasjon. I denne fasen mottar mange pasienter behandling på sykehus, dels for symptomlindring, dels for livsforlengende behandling og dels på grunn av livstruende komplikasjoner. Parallelt følges pasientene av fastlege og øvrig kommunal helse- og omsorgstjeneste, og eventuelt palliativt senter. For fastlegen er det i denne fasen viktig å kjenne til symptomer knyttet til alvorlige komplikasjoner ved sarkom, som i all hovedsak er de samme som ved annen utbredt kreftsykom. Palliasjon og omsorg ved livets slutt for kreftpasienter må for øvrig tilpasses den enkeltes kliniske situasjon. Se også «Nasjonalt handlingsprogram med retningslinjer for palliasjon i kreftomsorgen» (Nasjonalt handlingsprogram for palliasjon i kreftomsorgen: nasjonal faglig retningslinje, 2019).
Forløpstider
Sist faglig oppdatert: 28.02.2022
Fra 1. september 2015 ble Pakkeforløp for sarkom innført. Da ble tidligere forløpstider erstattet av de nye tidene i Pakkeforløp for sarkom.
Om Pakkeforløp for kreft
Sist faglig oppdatert: 28.02.2022
Pakkeforløp for kreft skal gi forutsigbarhet og trygghet for pasient og pårørende, og er et standard pasientforløp som beskriver organisering av utredning og behandling, kommunikasjon/dialog med pasient og pårørende, samt ansvarsplassering og konkrete forløpstider. Pakkeforløpet starter når et helseforetak eller privat ideelt sykehus mottar en henvisning med begrunnet mistanke om kreft, eller når helseforetaket selv starter utredning med begrunnet mistanke om kreft.
Formålet med Pakkeforløp for kreft er at kreftpasienter skal oppleve et godt organisert, helhetlig og forutsigbart forløp uten unødvendig ikke-medisinsk begrunnet forsinkelse i utredning, diagnostikk, behandling og rehabilitering.
Forløpstidene i pakkeforløpet beskriver den maksimale tiden de ulike fasene i forløpet bør ta. Forløpstidene angis i kalenderdager. De enkelte fasenes forløpstid legges til slutt sammen til en samlet forløpstid, som angir tiden fra henvisning er mottatt til behandling er startet. Med utgangspunkt i pakkeforløpet skal et individuelt forløp tilrettelegges for hver enkelt pasient.
De regionale helseforetakene har det overordnede ansvaret for å sikre at pakkeforløpene med forløpstider blir implementert og fulgt opp.
Forløpstidene er normerende og ikke en pasientrettighet. Fortsatt er det lovmessige grunnlaget pasientrettighetsloven § 2‑2 (Lov om pasient- og brukerrettigheter (pasient- og brukerrettighetsloven)) og forskrift om prioritering av helsetjenester (Forskrift om prioritering av helsetjenester, rett til nødvendig helsehjelp fra spesialisthelsetjenesten, rett til behandling i utlandet og om klagenemnd (prioriteringsforskriften)). Av og til vil det av faglige grunner være noen pasienter som ikke kan utredes ferdig innen normert forløpstid for oppstart av første behandling. Årsaker til avvik fra de normerte forløpstidene bør dokumenteres i pasientjournalen.
Forløpstider for sarkom
Sist faglig oppdatert: 28.02.2022
I Pakkeforløp for sarkom er det utarbeidet følgende forløpstider:
| Forløpsbeskrivelse | Forløpstid | |
|---|---|---|
| Fra henvisning mottatt til første fremmøte utredende avdeling | 8 kalenderdager | |
| Fra første fremmøte i utredende avdeling til avsluttet utredning (beslutning tas) | 21 kalenderdager | |
| Fra avsluttet utredning til start behandling | Kirurgisk behandling | 14 kalenderdager |
| Fra avsluttet utredning til start behandling | Medikamentell behandling | 14 kalenderdager |
| Fra avsluttet utredning til start behandling | Strålebehandling | 14 kalenderdager |
| Fra henvisning mottatt til start behandling | Kirurgisk behandling | 43 kalenderdager |
| Fra henvisning mottatt til start behandling | Medikamentell behandling | 43 kalenderdager |
| Fra henvisning mottatt til start behandling | Strålebehandling | 43 kalenderdager |
Pakkeforløp for sarkom finnes omtalt på Helsedirektoratets nettsider.
Se www.helsedirektoratet.no.
Det er utarbeidet egne diagnoseveiledere for fastleger for inngang til pakkeforløp. Diagnoseveileder finnes på www.helsedirektoratet.no.
Diagnostikk og utredning
Symptomer og funn
Sist faglig oppdatert: 28.02.2022
Bløtvevssarkomer i ekstremitet og trunkus debuterer ofte som en synlig eller palpabel – og oftest voksende – hevelse uten smerter. Bensarkomer debuterer derimot oftest med smerter, mens palpasjonsfunnene kan være sparsomme. Sarkom gir sjelden forandringer i vanlige laboratorieprøver. Siden sarkom er sjeldent, vil symptomer fra muskel-skjelettsystemet i de aller fleste tilfeller ha en annen årsak. Imidlertid har en studie fra Århus i Danmark vist at én av tre pasienter som har sarkom ikke hadde de typiske alarmsymptomene før diagnosen (Dyrop et al., 2014).
Sarkom i retroperitoneum og indre organer gir ofte ingen eller diffuse plager, og kan bli store før de oppdages.
De ovennevnte faktorer gjør at diagnostikk av sarkom er komplisert og ressurskrevende. Det er nødvendig å øke bevissthet og kunnskap om sarkom i primærhelsetjeneste og i spesialisthelsetjeneste.
Bløtvevssvulster i ekstremitet og trunkus
Bløtvevssvulster kan forekomme i alle aldre. Det vanligste er at pasienten merker en kul, men oftest ingen smerter. Smerter oppstår først hvis tumor blir så stor at den påvirker omgivende vev, som nerver eller blodårer. Svulster som sitter dypt i låret eller i seteregion kan bli store før de oppdages.
Det kan være vanskelig å skille klinisk mellom overfladiske og dype svulster. En dyp svulst vil kunne føles hardere når omgivende muskel kontraheres. En overfladisk svulst vil ofte kunne beveges i forhold til underliggende fascie og muskulatur selv når muskelen kontraheres. En malign svulst vil ofte være hardere enn en benign, men dette er ikke noen absolutt sannhet. Både maligne og benigne svulster kan vokse. Man kan altså klinisk ikke skille mellom maligne og benigne svulster.
Bensvulster
Smerter er det vanligste symptom ved bensarkom, men symptomene kan i begynnelsen være vage, og ofte mangler spesifikke funn ved første kliniske undersøkelse. Smertene tiltar gjerne etter hvert som svulsten vokser, og går da gjerne fra å være belastningsrelaterte til å bli hvile- og nattesmerter. Det er ikke sjelden at disse pasientene har gått med smerter i lang tid før diagnosen blir stilt. Smertene oppfattes ofte som «voksesmerter», senebetennelser eller idrettsskader. Smerter som ikke går over i løpet av noen uker bør utredes videre med røntgen.
Hos noen pasienter er CRP forhøyet, spesielt ved Ewing sarkom, og dette kan mislede kliniker til å tro at pasienten har osteomyelitt.
Knokkelstrukturen der et bensarkom sitter blir svekket og det kan oppstå brudd ved minimale traumer (patologisk fraktur). Bensarkomer kan noen ganger debutere med en slik patologisk fraktur. Det er angitt at dette skjer hos cirka 15–20 % av pasientene med maligne benlesjoner (Salunke et al., 2014). Patologiske brudd må utredes med tanke på bakenforliggende sykdom før operativ behandling av bruddet.
Abdominalt og retroperitonealt sarkom
Sarkom i retroperitoneum og indre organer gir ofte ingen eller diffuse plager, og kan bli store før de oppdages. Ved retroperitonale sarkomer skyldes symptomene ofte tumors størrelse (median størrelse 15-20 cm) (Bonvalot et al., 2009; Gronchi et al., 2009; Toulmonde et al., 2014). I flere tilfeller oppdages tumor som en oppfylning av pasienten selv, eller som tilfeldig funn ved legebesøk (P. W. T. Pisters, 2002; Stoldt & Geraghty, 1999). Omkring en fjerdedel av GIST oppdages som tilfeldig funn på CT eller ved endoskopi (Mucciarini et al., 2007). Gastrointestinal blødning sees hyppig, og tumor kan gi seg til kjenne som en palpabel tumor. Dette gjelder alle abdominale sarkomer. Akutt abdomen (smerter, peritonitt, ileus) som debutsymptom forekommer, men er ikke vanlig. Almennsymptomer og vekttap er ofte uttrykk for disseminert sykdom, som er tilstede ved diagnose hos 10-20 % av pasienter med GIST (Emile et al., 2012).
Gynekologisk sarkom
Symptomene ved gynekologisk sarkom er uspesifikke og vanskelige å skille fra symptomene ved benigne tilstander. Blødningsforstyrrelser forekommer hos en del pasienter (Abeler et al., 2009; Nordal, 1998).
- Menoragi (10 %)
- Meno/metroragi (20 %)
- Postmenopausal blødning (40 %)
- Abdominalsmerter
- Utspilt abdomen
- Urinveissymptomer
Sarkomutvikling i myomatøs uterus skal mistenkes ved:
- Vekst av «myomer» hos postmenopausale kvinner
- Hurtig vekst, selv om «hurtigvoksende myom» ikke er typisk for sarkom og bare ble funnet hos 1 av 371 pasienter (0,27 %) med slik tilstand (Parker, Fu, & Berek, 1994).
- Tumor i uterusveggen med uskarp avgrensning og/eller innvekst i parametrium bedømt ved ultralyd/CT/MR.
Henvisningsrutiner
Sist faglig oppdatert: 28.02.2022
Bløtvevssvulster i ekstremiteter og trunkus
Bløtvevssarkom kan forveksles med et stort antall benigne tilstander, både av pasienten selv og av lege. Riktig og rask behandling vil avhenge av at de maligne svulstene siles ut ved klinisk undersøkelse og bildediagnostikk, og henvises urørt videre. For å oppnå dette, er det nødvendig å vurdere og utrede en rekke tilstander som i ettertid viser seg å være benigne. Forsøk på å fjerne svulsten eller å ta biopsi kan føre til kontaminering av involverte strukturer. Dette vil kunne komplisere senere kirurgi, og redusere muligheten for optimal behandling.
MR er den beste bildediagnostiske metoden for utredning av bløtvevssvulster (M.J. Kransdorf & Murphey, 2000, 2006). Det er indikasjon for MR ved dype svulster, ved subkutane svulster som ved palpasjon er > 5 cm (en tumor som klinisk måles til 5 cm, vil på MR oftest være ca. 4 cm), ved klinisk vekst av uavklarte overfladiske lesjoner, selv om lesjonene er små, og ved klinisk malignitetssuspekte svulster.
MR er velegnet til å identifisere typiske lipomer. Subkutane lipomer diagnostisert med MR trenger oftest ingen behandling, men kan uavhengig av størrelse behandles ved lokalsykehus eller privat kirurg uten ytterligere vevsdiagnostikk dersom det er klinisk indikasjon. Tilfeldig påviste inter-/intramuskulære lipomer som ikke gir symptomer trenger heller ikke henvises til sarkomsenter. Ved symptomgivende dype lipomer, lipomer med dokumentert vekst eller ved behov for teknisk assistanse på grunn av størrelse/lokalisasjon bør pasienter henvises for vurdering ved sarkomsenter.
Pasienter med dyptliggende svulster som ikke er lipom skal henvises til sarkomsenter og bildene vurderes der, uansett MR-funn. MR kan identifisere de subkutane svulstene under 5 cm som ikke representerer lipomer. MR-undersøkelsen skal alltid vurderes ved sarkomsenter selv om tumor er liten.
MR-undersøkelsens diagnostiske verdi avhenger av teknisk kvalitet, undersøkelsesprotokoll med relevante serier, og riktig tolkning av bildene. Siden bløtvevssarkomene er sjeldne og kan ha variabelt radiologisk utseende, er det risiko for feiltolkning med tenke på andre tilstander enn malign tumor (cyste, hematom, muskelruptur, betennelse, abscess, godartet svulst osv. (Berquist, Ehman, King, Hodgman, & Ilstrup, 1990; M.J. Kransdorf & Murphey, 2000, 2006; Moulton et al., 1995). Terskelen må derfor være lav for henvisning videre til tverrfaglig vurdering ved uklare MR-funn. Utenfor sarkomsenter er det viktigst å gjøre tilstrekkelige undersøkelser for å oppdage tumor (MR er ofte tilstrekkelig. Biopsi skal ikke utføres). På sarkomsenter vil behovet for ytterligere bildediagnostikk og vevsdiagnostikk vurderes, og undersøkelsene skreddersys etter tumors lokalisasjon og hvilken type behandling som er aktuell.
Anbefalingene om MR-diagnostikk har erstattet tidligere kliniske retningslinjer for hvilke pasienter med ekstremitets- og trunkuslokaliserte svulster som skal henvises til sarkomsenter uten forutgående biopsi eller kirurgi. Disse retningslinjene er i tråd med internasjonal konsensus (Bauer et al., 2001; Bhangu, Beard, & Grimer, 2004; Dangoor et al., 2016; Gustafson, Dreinhofer, & Rydholm, 1994; Rydholm, 1998) som er vedtatt av Skandinavisk sarkomgruppe (SSG):
- Alle dype svulster uansett størrelse.
- Alle subkutane svulster klinisk > 5 cm.
- Svulster med annen grunn til malignitetsmistanke (tilbakefall etter tidligere kirurgi, rask vekst eller smerter).
Anbefalinger:
- MR anbefales ved dypt beliggende svulster, ved subkutane svulster som ved palpasjon er > 5 cm, ved klinisk vekst av uavklarte overfladiske lesjoner, selv om lesjonene er små, og ved klinisk malignitetssuspekte svulster.
- Pasienter med bløtvevssvulster som ikke er lipom skal henvises til sarkomsenter og bildene vurderes der, uansett MR-funn.
- Pasienter skal henvises uten forutgående biopsi.
Bensvulster
Siden bensarkomer er sjeldne, vil symptomer og funn fra muskel-skjelettsystemet i de aller fleste tilfeller ha annen årsak. Diagnosen kan derfor ikke stilles på bakgrunn av klinikk alene, men avhenger av at det rekvireres relevant bildediagnostikk. Ved smerter fra skjelettet (trunkus, ekstremiteter), palpabel kul og/eller annen suspekt klinikk, skal det være lav terskel for henvisning til røntgenundersøkelse. Ved negativt røntgenfunn og persisterende symptomer må det gjøres ny klinisk vurdering. Røntgen bør gjentas og eventuelt suppleres med MR.
Bensarkom skal mistenkes og pasienter skal henvises til sarkomsenter ved (C. Gerrand et al., 2016; Grimer, Carter, Tillman, & Abudu, 2001; Grimer & Sneath, 1990; Widhe & Widhe, 2000):
- Suspekte radiologiske funn, som bentap, destruksjon av cortex, periostreaksjon eller bennydannelse.
- Mistanke om patologisk fraktur (uten at det foreligger kjent kreftdiagnose med spredning til skjelett).
- Uklar diagnose og mistanke om bensvulst.
En rekke patologiske tilstander i ben kan ligne bensarkom. I mange tilfeller kan diagnosen avklares ved at bildene vurderes ved sarkomsenter sammen med relevante kliniske opplysninger. Benigne tilstander kan i mange tilfeller deretter tas hånd om av primærhelsetjenesten eller lokalsykehus.
Mange pasienter henvises til MR uten forutgående røntgenundersøkelse. Til tross for økende tilgjengelighet på MR-undersøkelser kan det være lang ventetid. Røntgen er imidlertid den enkleste, minst ressurskrevende og lettest tilgjengelige undersøkelsen for påvisning av patologi i skjelettet. Ved mange benlesjoner vil røntgen være den avgjørende modaliteten i diagnostisk avklaring (Miller, 2008; Moser & Madewell, 1987).
De fleste bensarkom er synlig på røntgen når tumor gir symptomer, men kan overses eller forveksles med benigne tilstander. Standard MR-protokoller som benyttes ved generell utredning av ben- og leddsmerter er mer spesifikt innrettet på leddundersøkelse og sjelden tilstrekkelige til tumordiagnostikk. Det er dermed flere kilder til forsinket diagnostikk, både klinisk og radiologisk.
Ved mistanke om bensarkom er det mest hensiktsmessig at utredningen gjøres ved, eller etter konferanse med, det sykehuset som skal behandle pasienten. Biopsi skal planlegges i samarbeid med den ortopeden som skal utføre det endelige kirurgiske inngrepet, siden biopsikanalen som hovedregel skal fjernes sammen med tumor. Det er vesentlig at man unngår å kontaminere de strukturer som skal brukes til rekonstruksjon eller er viktige å bevare av annen grunn (Mankin, Lange, & Spanier, 1982). Pasienten skal derfor henvises uten forutgående biopsi.
Anbefalinger:
- Ved klinisk mistanke om mulig bensvulst skal pasienter utredes med røntgen og eventuelt MR, og det skal være lav terskel for å sende bilder til vurdering ved sarkomsenter.
- Ved mistanke om bensarkom skal pasienten henvises til Flerregional behandlingstjeneste for kirurgi ved bensarkom ved Oslo universitetssykehus eller Haukeland universitetssykehus.
- Pasienten skal henvises uten forutgående biopsi.
Abdominalt og retroperitonealt sarkom
Kliniske og radiologiske funn er bakgrunn for videre henvisning til sarkomsenter. Ved mistanke om sarkom skal pasienten henvises uten forutgående biopsi eller operasjon. Pasienter med mistenkt eller fastslått abdominalt eller retroperitonealt sarkom skal ikke opereres på steder uten kirurgisk erfaring og kompetanse ADDIN EN.CITE.DATA (Casali et al., 2022; Gronchi et al., 2021).
Gynekologisk sarkom
Ved klinisk eller radiologisk mistanke om gynekologisk sarkom skal pasienten henvises sarkomsenter uten forutgående biopsi eller operasjon.
Anbefalinger:
- Ved mistanke om retroperitonealt, abdominalt eller gynekologisk sarkom skal pasienten utredes med CT og eventuelt MR, og hvis mistanken ikke avkreftes skal man henvise videre til sarkomsenter.
- Pasienten skal henvises uten forutgående biopsi.
Sarkom hos barn
Utredning av sarkom hos barn bør skje ved barne-/ungdomsavdelinger og i nært samarbeid med sentre med spesialkompetanse i håndtering av sarkom. Det er viktig at utredningen skjer i henhold til gjeldende protokoller. Det vises til «Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn» (Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn: nasjonal faglig retningslinje, 2020).
Bildediagnostisk utredning
Sist faglig oppdatert: 28.02.2022
Radiologisk og nukleærmedisinsk utredning av bløtvevssvulster i ekstremitet og trunkus
Det mangler eksakte radiologiske kriterier for å skille mellom maligne og benigne bløtvevssvulster (Gielen et al., 2004; M.J. Kransdorf & Murphey, 2006; Ma, McCarthy, Bluemke, & Frassica, 1998; Moulton et al., 1995; Weatherall, 1995), bortsett fra lipomer som kan identifiseres ved adekvat utført MR. Erfarne radiologer kan i mange tilfeller gi en godt begrunnet tentativ diagnose, men sikker diagnose forutsetter like fullt vevsdiagnostikk. Alle bløtvevssvulster som ikke er lipomer skal som hovedregel gjennomgå vevsdiagnostikk før kirurgi. Uavhengig av tentativ diagnose skal dype og store svulster alltid henvises videre i henhold til kriteriene beskrevet under kapittel Henvisningsrutiner.
MR er den overlegne metoden i utredning av bløtvevssvulster (M.J. Kransdorf & Murphey, 2000, 2006; Petasnick, Turner, Charters, Gitelis, & Zacharias, 1986). Andre metoder er aktuelle som supplement eller ved kontraindikasjon mot MR.
Ultralyd: God oppløselighet i overfladisk bløtvev, men skiller dårlig mellom vevstyper i tumor og er ikke tilstrekkelig som eneste utredning av sarkom. Kan brukes for utredning dersom det er usikkerhet om det foreligger tumor, og for kartlegging av overfladisk antatt benigne tumorer under 5 cm (Griffith et al., 2020; E. H. Y. Hung et al., 2020; Lakkaraju, Sinha, Garikipati, Edward, & Robinson, 2009; Rowbotham, Bhuva, Gupta, & Robinson, 2012). Dersom det ikke kan stilles sikker og benign diagnose ved ultralyd må MR gjøres. Ultralyd er viktig som veileder ved biopsi. Generelle retningslinjer for biopsi må følges.
Røntgen: Gir lite informasjon om bløtvev, men kan være aktuelt ved store og dype svulster på ekstremitetene, og for å kartlegge benaffeksjon. Gir pekepinn om spesifikk diagnose i enkelte tilfeller, f.eks. flebolitter ved hemangiom/vaskulær malformasjon og ringformet forbening ved myositis ossificans (M.J. Kransdorf & Murphey, 2000).
Angiografi: Gir sjelden tilleggsinformasjon av betydning for behandlingsvalg. Angiografi er dårligere enn MR for fremstilling av tumors relasjon til kar (viser lumen, ikke ytre del av karveggen), og gir dårlig fremstilling av tumormarginer. Angiografi har ingen plass i rutineutredningen, kun etter individuell vurdering ved konkret problemstilling (for eksempel vurdering av embolisering før operasjon).
CT: Benyttes når MR ikke er tilgjengelig, eller ved kontraindikasjon mot MR. Kan gi tilleggsinformasjon i utvalgte tilfeller og i spesielle lokalisasjoner etter individuell vurdering.
MR: Ved bløtvevssvulster er god MR-undersøkelse en nødvendig forutsetning for planlegging av videre behandling (M.J. Kransdorf & Murphey, 2000, 2006). Fremstilling av tumors nøyaktige anatomiske lokalisasjon er nøkkelen til tumorfri margin ved kirurgi. MR kan vise utbredelse i forhold til kar/nerver, omgivende bløtdeler, ben og ledd. God bløtdelsfremstilling kan i de fleste tilfeller differensiere mellom viabelt tumorvev, cystiske områder, reaktive forandringer (hyperemi/ødem), nekrose og blødning, og styre biopsi til de områder av tumor hvor det er størst sjanse til å få representativt materiale.
Forutsetningen for å kunne bruke MR-undersøkelse til tumordiagnostikk er at den er teknisk adekvat og inneholder relevante sekvenser. Det finnes et meget stort spekter av mulige pulssekvenser. Hjørnesteinen er fortsatt de basale konvensjonelle spin ekko T1 og væskesensitive sekvenser i konvensjonelle anatomiske plan. Undersøkelsen må som et minimum omfatte de basale sekvenser for vevskarakteristikk og anatomisk oversikt.
Erfaringsmessig er svært mange MR-undersøkelser gjort utenfor tumorsenter av utilfredsstillende kvalitet for sarkomdiagnostikk. Det kan dreie seg om for dårlig oppløsning i bildene for vurdering av detaljer, for stort eller for lite synsfelt, eller sekvensvalg som ikke tillater differensiering mellom fett og vann, cyste og solid tumor. Dette til tross for at det ofte tas svært mange serier.
Som basal tumorprotokoll anbefales T1 og STIR (eller annen vannsensitiv, fettsupprimert serie) oversiktserier med stor FOV i lengdeplan; vanligvis coronal, eventuelt sagittal, hvis det vurderes som bedre egnet ut fra tumors beliggenhet. Spin ekkobasert T1- og T2 uten fettsuppresjon i aksialplan med mindre FOV, men dekkende aktuelle lesjon og de samme snitt T1-vektet med fettsuppresjon og i.v. kontrast (M.J. Kransdorf & Murphey, 2000, 2006). Disse fem seriene er ofte tilstrekkelig som preoperativ utredning. Funksjonell MR blir stadig mer benyttet i tumorutredning. Dynamiske kontrastserier med perfusjon, MR-angiografi og diffusjon erstatter ikke morfologiske serier, men er kun supplerende opptak som eventuelt kan gi verdifull tilleggsinformasjon om vaskularisering og cellularitet, og om behandlingsrespons.
Som konklusjon er god anatomisk fremstilling og vevsdifferensiering det viktigste ved MR av bløtvevssvulster. MR kan ikke stille histologisk diagnose, og i de fleste tilfeller må bildeutredningen kompletteres med nålebiopsi.
FDG PET/CT: Over 90% av bløtvevssarkomer er rapportert å være FDG-avide (Roberge et al., 2012), og høygradige sarkomer har som oftest høyere opptaksintensitet enn lavgradige (Fendler et al., 2015; Macpherson et al., 2018; Rakheja et al., 2012). Ved heterogene svulster antas det at områder med høyt opptak representerer den mest aggressive delen av tumor, og at biopsi fra slike områder er mest representative. Det er rapportert at høy opptaksintensitet predikerer dårlig prognose (Becher & Oskouei, 2015; Chen et al., 2017; Fuglø, Jørgensen, Loft, Hovgaard, & Petersen, 2012; Kubo, Furuta, Johan, & Ochi, 2016; Y. J. Li, Dai, Cheng, Zhang, & Tu, 2016; Palmerini et al., 2017). FDG PET/CT kan også være nyttig i responsevaluering (Lim, Johnny Ong, Tan, & Ching Teo, 2019; Macpherson et al., 2018), men forutsetter at det er utført PET før oppstart behandling. FDG PET kan således gi tilleggsinformasjon i primærutredning av bløtvevssarkomer på flere måter, men klinisk nytteverdi er ikke presist kartlagt.
Radiologisk og nukleærmedisinsk utredning av abdominalt og retroperitonealt sarkom
En teknisk god CT thorax/abdomen/bekken med intravenøs kontrast i portovenøs fase vil som regel være tilstrekkelig radiologisk utredning for retroperitoneale svulster. Differensialdiagnoser er nyrekarsinom, binyretumor, benign nerveskjedetumor, germinalcelletumor, malignt lymfom, karsinommetastase, reaktive tilstander og betennelser m.m. Funksjonen av gjenværende nyre må vurderes hvis det planlegges nefrektomi. Ved svulster i lyske/skrotum må hele retroperitonealrommet utredes.
Ved abdominale svulster er CT-abdomen/bekken med intravenøs kontrast standard, men kan erstattes med MR. Lungemetastaser er ytterst sjelden ved GIST, men lungene bør avklares ved diagnose. Øvre endoskopi er ikke påkrevet ved GIST i ventrikkel med karakteristisk billeddiagnostikk. Adenokarsinom vil kunne utelukkes på CT, gjerne utført som gastrografi (vann eller CO2 i ventrikkel). MR-bekken gjøres ved tumor i rektum. For FDG PET/CT, se avsnitt over, om Radiologisk og nukleærmedisinsk utredning av bløtvevssvulster i ekstremitet og trunkus.
Radiologisk og nukleærmedisinsk utredning av gynekologisk sarkom
Ved mistenkt eller påvist gynekologisk sarkom skal MR bekken og CT thorax/abdomen/bekken utføres. For FDG PET/CT, se se avsnitt over, om Radiologisk og nukleærmedisinsk utredning av bløtvevssvulster i ekstremitet og trunkus.
Metastaseutredning ved bløtvevssarkom
Ekstremitetslokaliserte sarkomer metastaserer som oftest hematogent til lunger. GIST metastaserer nesten utelukkende til lever og peritoneum (DeMatteo et al., 2000). Retroperitoneale sarkomer metastaser helst til lunger eller residiverer peritonealt (Gronchi et al., 2016). Enkelte tumortyper har større tendens til lymfeknutemetastaser (synovialt sarkom, rhabdomyosarkom), men generelt metastaserer sarkomer sjelden til lymfeknuter. Metastaseutredning ved sarkomer i ekstremiteter består av CT-thorax. Det bør være liberal indikasjon for CT-abdomen og bekken, spesielt ved tumor i underekstremiteter. For abdominale og retroperitoneale sarkomer er CT-thorax/abdomen/bekken standard.
FDG PET/CT: Sensitivitet for deteksjon av metastaser med FDG PET/CT helkroppsundersøkelse (fra vertex inkludert over- og underekstremiteter) er høy. En metaanalyse viser høy sensitivitet, spesifitet, PPV og NPV, henholdsvis 97%, 98%, 97% og 98%, for påvisning av metastaser fra sarkomer (Lim et al., 2019). En retrospektiv studie viser også at FDG PET/CT har høy sensitivitet (96%), positiv prediktiv verdi (96%) og negativ prediktiv verdi (99%) for påvisning av metastatatisk sykdom (Macpherson et al., 2018). Det anbefales at FDG/PET CT kan vurderes som ledd i metastaseutredning, spesielt ved høygradig maligne bløtvevssarkomer.
Radiologisk og nukleærmedisinsk utredning av bensvulster
Gullstandarden i utredningen av bensvulster i ekstremiteter er røntgen og MR, som kompletterer hverandre (Alyas, James, Davies, & Saifuddin, 2007; M. J. Kransdorf, 2009; Miller, 2008). Ved mange benigne lesjoner er røntgen tilstrekkelig utredning, mens MR alltid skal ledsages av konvensjonelt røntgenbilde. I andre lokalisasjoner enn ekstremiteter er CT ofte nødvendig i stedet for røntgen, se nedenfor.
Røntgen: Viser skjelettets kalkholdige komponent (knokkelstruktur) og annet kalkholdig vev, men ikke bløtvev og benmarg. Røntgen gir viktig informasjon om vekstmønster og reaksjon fra periost og benvev, og gir beste pekepinn om diagnose. Tidlig i forløpet av et bensarkom kan funnene være beskjedne. For å oppdage en tumor i så tidlig stadium som mulig, er det viktig at bildene granskes med denne muligheten for øyet. Av pasienter som får påvist en malign bentumor, har inntil en fjerdedel tatt et røntgenbilde tidlig i forløpet som er beskrevet negativt, men der tumor retrospektivt var til stede (Grimer et al., 2001; Grimer & Sneath, 1990). Bildene må spesifikt granskes med henblikk på uregelmessig knokkelstruktur og avgrensning, periostreaksjon, erosjon eller destruksjon av cortex, bløtdelshevelse med eller uten kalk.
CT: Bedre alternativ enn røntgen for fremstilling av knokler som er vanskelige å vurdere på røntgenbilder (bekken, sakrum, sternum, virvler). Viser kortikalt gjennombrudd og detaljer i knokkelstruktur, samt utseende og eksakt lokalisasjon av forkalkninger og forbeninger. Dette er spesifikk tilleggsinformasjon til MR (Aisen et al., 1986; Tehranzadeh, Mnaymneh, Ghavam, Morillo, & Murphy, 1989). Indikasjon vurderes individuelt.
Når CT er supplement til MR, er det oftest tilstrekkelig med benalgoritme og serie uten intravenøs kontrast. Hvis MR ikke utføres (på grunn av kontraindikasjon eller andre forhold), må CT også gi svar på utbredelse i benmarg, omgivende bløtdelstumor, vaskularisering og relasjon til nerver/kar/muskulatur/fascie. Det krever bilder uten og med kontrast, eventuelt i flere faser, og rekonstruksjon i flere plan.
CT-veiledet biopsi er godt egnet ved benlesjoner. Generelle retningslinjer for biopsering må følges.
MR: MR fremstiller skjelettets bløtdelskomponent og er ved adekvat sekvensvalg alle andre metoder overlegen i bedømmelse av benmarg (Aisen et al., 1986; Alyas et al., 2007). Stor kontrast mellom normal fettholdig benmarg og tumorvev (T1-vektede bilder) gir utmerket fremstilling av tumorutbredelsen i margrommet, uavhengig av om trabekelverket er destruert. Kan påvise skip-metastaser (separat tumormanifestasjon i samme margrom som hovedtumor) og eventuell intraartikulær tumorutbredelse. Coronal- og sagittalplan er best for vurdering av utbredelse og lokalisasjon i knokkelens lengderetning, mens aksialplan er best for å skille anatomisk mellom tumor, ben, bløtvev (muskulatur, subkutis, hud) og kar/nerver.
MR viser prinsipielt ikke knokkelstruktur eller bendestruksjon, bare indirekte, og er lite sensitiv og spesifikk for forkalkninger.
Ved mistanke om osteogent sarkom eller Ewing sarkom skal hele margrommet i lange rørknokler fremstilles, samtidig som det er nødvendig med best mulig detaljfremstilling av tumorområdet.
Samme basisprotokoll anbefales som for utredning av bløtvevssvulster (M.J. Kransdorf & Murphey, 2000): Coronal (evt. sagittal) T1 og STIR. Aksial T2, Aksial T1 (uten fettsuppresjon, viktig for å identifisere fettsjikt). Aksial T1 med fettsuppresjon etter kontrast. Ved utredning på sarkomsenter må MR-protokollen tilpasses individuelt. Nøyaktig serievalg vil avhenge av tumors type og beliggenhet, og egenskaper ved MR-maskinen.
Angiografi: Sjelden aktuelt.
Ultralyd: Viktigst som veileder for biopsi. Ved benlesjoner forutsetter ultralydveiledet biopsi at det foreligger bløtdelskomponent eller defekt i cortex inn mot lesjonen. Riktig biopsitilgang vurderes på forhånd i hvert enkelt tilfelle i samråd med ortoped/kirurg.
FDG PET/CT: Konvensjonelle osteosarkomer og Ewing sarkom har gjennomgående høyt opptak av FDG (Huang et al., 2018; F. Liu, Zhang, Zhou, & Dong, 2019). Mulig nytteverdi for FDG PET/CT er de samme som ved bløtvevssarkom. PET kan gi veiledning når det gjelder hvilket område av tumor som bør biopseres (Expert Panel on Musculoskeletal et al., 2020), høy opptaksintensitet er assosiert med dårlig prognose (Huang et al., 2018; Hwang et al., 2016; Kubo et al., 2016) og PET kan brukes som responsevaluering (Hongtao et al., 2012; Lim et al., 2019; Macpherson et al., 2018). FDG PET/CT kan således gi tilleggsinformasjon ved primærutredning av bensarkomer på flere måter, men klinisk nytteverdi er ikke presist kartlagt.
Metastaseutredning ved bensarkom
Bensarkomer metastaserer som oftest hematogent. Osteosarkomer og kondrosarkomer metastaserer oftest til lunge, med skjelett som den nest vanligste lokalisasjonen. Ved Ewing sarkom sees oftere spredning til andre organer, selv om lunge og skjelett også her er det vanligste. Metastaseutredning består alltid av CT-thorax, med lav terskel for å supplere med CT- abdomen/bekken. Ved konvensjonelt osteosarkom og Ewing sarkom anbefales FDG PET/CT staging (ACR apropriateness criteria) (Expert Panel on Musculoskeletal et al., 2020; Huang et al., 2018; F. Liu et al., 2019). MR totalcumna og bekkenskjelett kan vurderes, spesielt dersom det ikke utføres FDG PET/CT. NaF PET/CT, eller skjelettscintigrafi med SPECT/CT, bør utføres ved osteosarkom for å oppdage skjelettmetastaser. NaF PET viser osteoblastaktivitet som skjelettscintigrafi, men er langt mer sensitiv på grunn av høyere bildeoppløselighet. Skjelettscintigrafi med SPECT/CT har lavere sensitivitet, men kan utføres dersom NaF PET/CT ikke er tilgjengelig.
Anbefalinger:
- MR er den foretrukne metoden til påvisning og utredning av bløtvevssvulster i ekstremiteter, og er vesentlig i preoperativ utredning av bløtvevssarkom her. Ved kontraindikasjon mot MR gjøres CT.
- CT med intravenøs kontrast er standard ved mistanke om abdominalt/retroperitonealt sarkom.
- CT, røntgen, ultralyd og angiografi kan være aktuelle som supplement til MR etter individuell vurdering.
- Ultralyd kan påvise tumor og være veileder for biopsi, men er utilstrekkelig som eneste preoperative bildefremstilling av bløtvevssarkom.
- Generell metastaseutredning ved bløtvevssarkom består av CT-thorax, eventuelt supplert med CT-abdomen/bekken ved sarkom i ekstremiteter og CT- thorax/abdomen/bekken ved abdominalt/retroperitonealt sarkom. FDG PET/CT kan vurderes, spesielt ved høygradig maligne svulster.
- Røntgen er den primære undersøkelsen til påvisning og utredning av bensvulster i ekstremitet. Bensvulster i trunkus krever oftest CT i stedet for røntgen.
- MR er aktuelt som supplement ved bensvulster, og er nødvendig i preoperativ utredning av bensarkom. Hele margrommet skal dekkes ved mistenkt osteosarkom og Ewing sarkom, for påvisning av skip-metastaser. Ved kontraindikasjon mot MR gjøres CT.
- Generell metastaseutredning ved bensarkom består av CT-thorax og FDG PET/CT. NaF PET/CT eller skjelettscintigrafi bør utføres ved osteosarkom.
Biopsi
Sist faglig oppdatert: 28.02.2022
Dersom man ikke kan stille en sikker diagnose etter radiologisk og klinisk undersøkelse av pasienten, skal svulsten biopseres. Biopsitilgang ved grovnålsbiopsi eller åpen biopsi må først avklares med kirurg ved sarkomsenter. Biopsikanalen skal plasseres slik at den ikke unødig berører viktige kar, nerver eller muskelgrupper. Ved biopsi er det fare for kontaminasjon av kreftceller i biopsikanalen. Hvorvidt dette øker risikoen for et klinisk residiv, er usikkert (Robertson & Baxter, 2011). Risikoen er minimal ved biopsi av abdominale/retroperitoneale sarkomer (Berger-Richardson & Swallow, 2017; Eriksson et al., 2016; van Houdt et al., 2021). Ved ekstremitetslokaliserte svulster er det tradisjon å gjøre innstikket slik at biopsikanalen enkelt kan fjernes ved en senere kirurgisk reseksjon. Dette praktiseres ikke ved abdominale/retroperitoneale sarkomer. Biopsi kan tas veiledet av palpasjon, ultralyd, røntgengjennomlysning eller CT.
Ved gynekologiske sarkomer tas biopsi vanligvis ultralydveiledet transvaginalt i forbindelse med palpasjon i narkose, og eventuelt fraksjonert abrasio. Stikkanalen bør hvis mulig legges slik at denne kan eksideres ved etterfølgende operasjon.
Biopsi kan unnlates når svaret ikke får behandlingsmessig konsekvens, f.eks. ved helt entydig bildediagnostikk og der kirurgisk prosedyre vil være den samme uansett patologisvar.
Finnålspunksjon (FNAC) gjøres ved hjelp av en tynn nål og tar ut enkeltceller fra svulsten. FNAC kan utføres poliklinisk og uten bedøvelse. Den er best egnet for bløtvevssvulster eller bentumores med stor bløtdelskomponent, og gir materiale for cytologisk undersøkelse. Den cytologiske undersøkelsen har vist høy sensitivitet og spesifisitet og kan vanligvis skille mellom sarkom, metastaser, lymfom og plasmacelleneoplasi.
Grovnålsbiopsi kan gjøres ved både ben- og bløtvevssvulster. Ved grovnålsbiopsi får man ut en vevsylinder som viser vevsarkitektur, og ikke bare enkeltceller som ved FNAC.
Åpen biopsi gjøres hovedsakelig ved bensvulster eller i tilfeller hvor FNAC eller grovnålsbiopsi ikke gir tilstrekkelig materiale. Åpen biopsi praktiseres ikke ved abdominale/retroperitoneale svulster der radikal kirurgi er mulig.
Anbefalinger:
- Nålebiopsi av svulsten tas når diagnose ikke kan stilles med sikkerhet radiologisk og når histologisk diagnose får konsekvenser for behandlingen.
- Biopsi bør utføres ved sarkomsenter.
Patologi
Sist faglig oppdatert: 28.02.2022
Sarkomer er sjeldne, og det er derfor vanskelig som patolog å få tilstrekkelig erfaring. Flere benigne svulster og reaktive lesjoner kan ligne på sarkom morfologisk. Diagnostikken bør derfor utføres av patologer med erfaring og interesse for sarkomdiagnostikk. Ideelt sett skal biopsier tas på et sarkomsenter der man stiller en integrert diagnose med morfologi, immunhistokjemi og molekylær patologi i kombinasjon med klinisk og radiologisk informasjon. Om prøven ikke er diagnostisert ved et sarkomsenter skal det vurderes å innkalle biopsimaterialet for regranskning. Det anbefales at patologen i sin besvarelse av sarkom, eller der sarkom kan mistenkes, gir råd om henvisning til et sarkomsenter.
Patologen bør være medlem og delta i det tverrfaglige sarkomteamet ved sykehuset. Her kan kasus diskuteres, og kompletterende informasjon om radiologiske og kliniske funn innhentes. Diagnostikken bør foregå i nært samarbeid med kliniker og radiolog. Ved vanskelige kasus bør preparater sendes for «second opinion» til andre patologilaboratorier i inn- eller utland.
Biopsier og operasjonspreparater bør så langt som mulig leveres ufiksert til patologilaboratoriet. Ferskt materiale er nødvendig for cytogenetisk undersøkelse. Vev anbefales frosset til biobank for å kunne utføre eventuelle molekylærpatologiske undersøkelser.
Remisseopplysninger
Ved utredning av ben- og bløtvevssvulster er den kliniske informasjonen viktig for den morfologiske vurderingen. Opplysninger om kjønn, alder, relevant sykehistorie – inkludert tidligere påvist kreft, og om kjemoterapi eller strålebehandling er gitt – radiologiske funn, lokalisasjon, dybde og stadium må fremgå. I tillegg må ønsker om tilleggsanalyser påføres.
Anbefalinger:
- Relevant klinisk informasjon må være angitt på remissen.
- Lokalisasjon og orientering av preparatet skal framgå i remisseopplysningene.
Makroskopisk bedømning og snittuttak
Makroskopisk undersøkelse av biopsier
Hvis det tas grovnålsbiopsi er første prioritet å ta av vev til morfologisk evaluering. Hvis det foreligger flere biopsier, anbefales det at man fryser ned vev for eventuelle molekylærpatologiske analyser. Cytogenetisk analyse kan være vanskelig på nålebiopsier, men er det tilstrekkelig materiale kan det sendes ufiksert vev til dette. For å sikre at biopsien er representativ kan det gjøres imprint eller frysesnitt. Ved cytologisk prøvetagning kan det tas av vev til både genetiske undersøkelser og flowcytometri.
Makroskopisk undersøkelse av operasjonspreparater
- Ufikserte operasjonspreparater skal umiddelbart leveres til patologilaboratoriet sammen med utfylt remisse. Hvis de ikke kan leveres ufiksert, fikseres de i formalin. Første prioritet er vev til morfologisk diagnostikk, men når det er mulig skal ufiksert tumorvev tas av for nedfrysing. Dette er til molekylær patologi. Ca. 1 cm3 tumorvev deles i mindre biter, ca. 0,2 cm store, og fryses i minst –70 °C.
- Det er viktig at preparatet er entydig orientert fra kirurgen. Operatøren skal orientere preparatet med suturer. Tegninger fra kirurgen, og radiologiske funn, er også til hjelp. Det anbefales at kirurg og patolog samarbeider om bedømmelsen av reseksjonsrender ved å inspisere det makroskopiske preparatet sammen.
- Fotografisk dokumentasjon anbefales.
- Det fikserte preparatet og tumor måles i tre dimensjoner.
- Beskriv typen vev rundt tumor.
- Reseksjonsrendene bedømmes på operasjonspreparatet etter fiksering i formalin. Tusjing av reseksjonsrendene anbefales.
- Preparater skjæres i ca. 1 cm tykke skiver, og snitt tas fra områder der marginen er minst. Den minste marginen skal måles, og typen vev skal beskrives. Prøv å angi mål på marginen proksimalt, kaudalt, anteriort, posteriort og til sidene.
- Tumors snittflate skal beskrives (konsistens, farge og blødninger). Andel makroskopisk tumornekrose skal angis i prosent.
Snittuttak
- Ta minst ett snitt per cm tumordiameter, for eksempel minst 6 snitt fra en 6 cm stor tumor. For svært store svulster er det vanligvis nok med 10 snitt fra lesjonen, men hvis tumor er heterogen er det viktig å ta snitt fra ulike områder.
- Snitt fra tumors avgrensing mot omgivelsene for å se på eventuell karinvasjon og vekstmåte.
- Snitt av nekrotisk vev og områder med blødninger.
- Snitt av kar i reseksjonsflaten i amputasjonspreparat.
- Snitt fra biopsikanalen.
- Snitt fra alle lymfeknuter.
- Storsnitt anbefales for å vurdere tumors heterogenitet, karinvasjon og vekstmåte.
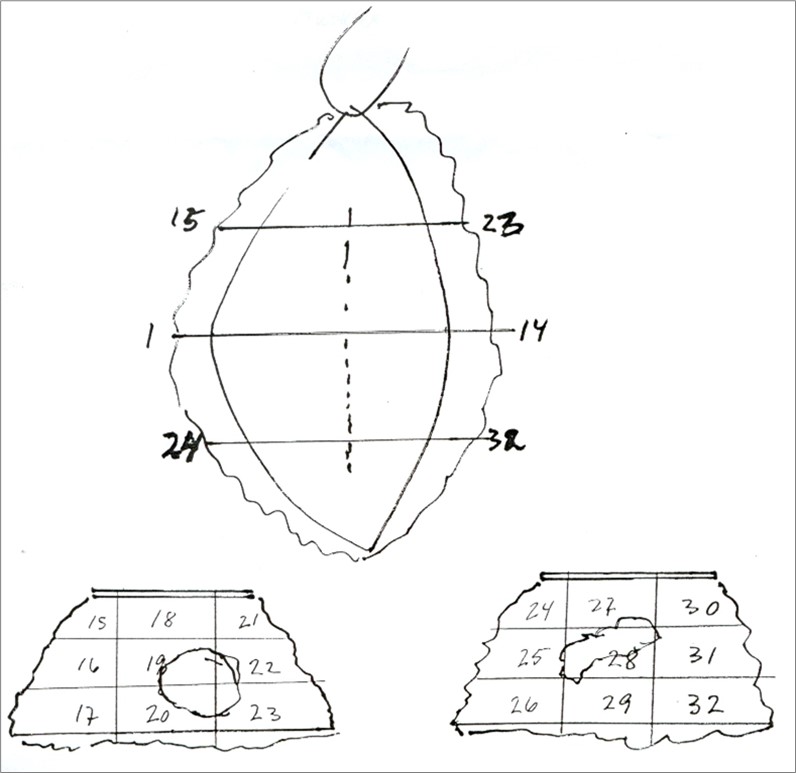
Eksempel på snittuttak:
Tumors dybde
Tumordybden skal angis både makroskopisk og mikroskopisk.
Kutan: Tumor starter i hud og kan infiltrere i subkutis.
Subkutan: Tumor er lokalisert i fettvevet mellom hud og den subkutane fascie. Hvis den infiltrerer inn i, eller gjennom fascien, skal den kategoriseres som dyp.
Dyp fascie: Den fascien som skiller subkutis fra underliggende skjelettmuskulatur.
Intramuskulær/intraossøs: Tumor er lokalisert under den dype fascie og har sitt opphav i muskulatur eller benvev, og er omgitt av muskelfascie/periost som ikke er infiltrert av tumor.
Ekstramuskulær/ekstraossøs: Tumor er lokalisert under den dype fascie og ligger mellom muskulatur, eller i området mellom muskulatur og benvev.
Ben- eller periostinfiltrasjon: Skal angis.
Anbefalinger:
- Ved FNA og grovnålbiopsier skal materiale til morfologisk undersøkelse prioriteres.
- Dersom det er tilstrekkelig materiale, anbefales det at det tas materiale til nedfrysing (biobank). Cytogenetisk analyse kan vurderes.
- Kompliserte operasjonspreparater anbefales vurdert av patolog og kirurg sammen.
Mikroskopisk undersøkelse og spesialundersøkelser
Mikroskopisk undersøkelse
Den mikroskopiske undersøkelsen av morfologi danner basis for den patologiske diagnosen, og det er den som avgjør hvilke spesialundersøkelser som er nødvendige.
Ben og bløtvevssvulster skal klassifiseres etter siste versjon av WHO-klassifikasjonen (WHO Classification of Tumours Editorial Board, 2020). Enzinger & Weiss «Soft tissue tumors» anbefales også (Goldblum et al., 2019).
Diagnosen baserer seg på morfologi, immunfenotyping og molekylær patologi. Ved bensvulster er det viktig å sette seg inn i den radiologiske undersøkelsen for å få eksakt kjennskap til lokalisasjon og vekstmønster.
Immunhistokjemiske undersøkelser
I sarkomdiagnostikken er det ofte nødvendig å gjøre immunhistokjemisk undersøkelse, ofte med et bredt panel av antistoffer. Paneler med antistoffer for ulike morfologiske grupper er nyttig, for eksempel for GIST, småcellete tumores og spolcellete tumores der relevante antistoffer, som modifiseres av kliniske opplysninger og morfologi, er inkludert. Dette kan gjøres i en seanse eller trinnvis. Disse paneler med antistoffer kan legges inn i systemene som benyttes i laboratoriene (LIMS).
Flowcytometriske undersøkelser
Flowcytometri er noen ganger av betydning som kompletterende undersøkelse ved utredning av småcellet tumor. Differensialdiagnosen malignt lymfom kan ofte raskt utelukkes med denne metoden.
Molekylærpatologiske undersøkelser
I sarkomdiagnostikken er det nødvendig med genetiske undersøkelser ved mange utredninger, spesielt mutasjonsanalyser og påvisning av translokasjoner. Eksempler på diagnoser der molekylære analyser har betydning som diagnostisk eller prediktiv markør er mutasjoner i KIT eller PDGFRA ved GIST, PAX3-FOXO1 eller PAX7-FOXO1 fusjoner ved rhabdomyosarkom, amplifikasjon av MDM2/CDK4 ved lavgradig og dedifferensiert liposarkom og mutasjon i CTNNB1 ved desmoid fibromatose. Mange histologiske subtyper og benigne differensialdiagnoser har patognomoniske translokasjoner, og påvisning av disse vil kunne ha diagnostisk og/eller prognostisk betydning
Det er i økende grad behov for diagnostiske molekylærgenetiske analyser, og konvensjonelle metoder (f.eks. Sanger sekvensering, RT-PCR og FISH) er arbeidskrevende å etablere for hver enkelt mutasjon eller translokasjon. Det anbefales derfor at annengenerasjons sekvenseringsteknologi er tilgjengelig ved patologiavdelinger som utfører sarkomdiagnostikk.
For informasjon om aktuelle genetiske funn, se WHOs klassifikasjon av ben og bløtvevssvulster og oppdatert litteratur på området. For informasjon om analyser og forsendelse, kontakt de aktuelle laboratorier.
Cytogenetiske undersøkelser
Ufiksert ferskt vev kan dyrkes for å påvise kromosomforandringer. Dette utføres ved Seksjon for kreftcytogenetikk, Radiumhospitalet, Oslo universitetssykehus. Materiale sendes på McCoy medium med utfylt remisse der det angis at det dreier seg om ben- eller bløtvevssvulst med mistanke om sarkom.
Materiale fra finnålsaspirasjon er som oftest ikke rikelig nok for cytogenetisk undersøkelse. Veiledning og transportmedium kan man få tilsendt ved henvendelse til Seksjon for kreftcytogenetikk, Radiumhospitalet, OCCI-bygget, Oslo universitetssykehus, telefon, 22 78 23 83.
Anbefalinger:
- Ben- og bløtvevssvulster skal klassifiseres etter siste versjon av WHO-klassifikasjonen. Immunhistokjemiske og molekylærpatologiske undersøkelser er viktig og nødvendig ved utredning av sarkomer. Det anbefales at annengenerasjons sekvenseringsteknologi er tilgjengelig ved patologiavdelinger som utfører sarkomdiagnostikk.
- Antistoffpaneler er nyttig ved utredning av sarkomer.
- Flowcytometri er nyttig for raskt å kunne skille enkelte sarkomer fra lymfomer.
Histopatologisk diagnose
En diagnose avgitt på et operasjonspreparat skal om mulig innholde:
| Histologisk diagnose | Basert på WHO-klassifikasjon |
|---|---|
| Tumors lokalisasjon og dybde |
|
| Tumors størrelse | Tre dimensjoner, mm |
| Malignitetsgrad | Bløtvevssarkom: Tregradig, FNCLCC Bensarkom: Tregradig. For GIST: Risikovurdering, angi hvilket risikovurderingssystem som brukes |
| Mitoser | Mitoser pr 10 HPF (standardiser etter FNCLCC-systemet) Ved GIST pr 5 mm2 |
| Nekrose | Ja, < 50 % Ja, ≥ 50 % Nei Kan ikke vurderes Eventuelt i henhold til aktuell protokoll. |
| Karinvasjon | Ja Nei Kan ikke vurderes |
| Vekstmåte | Diffust infiltrerende Bred front («pushing») Kan ikke vurderes |
| Type resektat med angivelse av hvilket vev som er affisert |
|
| Reseksjonsrender
| Vurdert som intralesjonelle eller frie. Angi avstand i mm fra tumor til reseksjonsflaten og typen vev. |
| Resultat av tilleggsanalyser | Immunhistokjemi Molekylær patologi Cytogenetisk analyse |
| Vurdering av histologisk behandlingsrespons ved preoperativ kjemoterapi og/eller strålebehandling | Gjelder først og fremst bensarkom som osteosarkom og Ewing sarkom. Følg retningslinjene i de aktuelle behandlingsprotokollene. |
Anbefaling:
- Relevant informasjon til klinikere skal komme fram i besvarelsen.
- Det anbefales å bruke diagnostiske maler.
Histologisk gradering av sarkom
Gradering av bløtvevssarkom
Det anbefales at det franske graderingssystemet for bløtvevssarkomer benyttes. Graderingssystemet FNCLCC (Fédération Nationale des Centres de Lutte le Cancer) er basert på tumors differensiering, mitosetall og nekrose og brukes på bløtvevssarkomer (WHO Classification of Tumours Editorial Board, 2020). Den totale poengsummen gir graden:
| Tumors differensiering | 1 poeng 2 poeng 3 poeng | Sarkom som ligner normalt og modent vev. Sarkom der histologisk type er sikker Embryonale og udifferensierte sarkomer. |
| Mitosetall | 1 poeng 2 poeng 3 poeng | 0–9 mitoser pr 10/HPF 10–19 mitoser pr 10/HPF ≥ 20 mitoser pr 10/HPF Et «high power field» (HPF) måler 0,1734 mm2. Dette medfører at mitosetallet må justeres i det enkelte mikroskop. |
| Tumornekrose | 0 poeng 1 poeng 2 poeng | Ingen nekrose < 50 % nekrose (også vurdert makroskopisk) ≥ 50 % nekrose (også vurdert makroskopisk) |
| Histologisk grad | Grad 1 Grad 2 Grad 3 | Totalt 2 eller 3 poeng Totalt 4 eller 5 poeng Totalt 6, 7 eller 8 poeng |
|
|
|
|
Gradering av bensarkom
Flere bensarkom gis ingen spesifikk maligntetsgradering da graden er gitt fra diagnosen. Dette gjelder for eksempel Ewing sarkom (grad 3), kordom (lavgradig malign) og dedifferensiert og mesenchymalt kondrosarkom (grad 3), se tabell nedenfor. For konvensjonelle kondrosarkom skal grad angis (grad 1-3).
| Grad | Type av sarkom i ben |
| |||
|---|---|---|---|---|---|
| 1 Lavgradig malign | Lavgradig sentralt osteosarkom Parostealt osteosarkom Klarcellet kondrosarkom |
| |||
| 2 Intermediær | Periostalt osteosarkom |
| |||
| 3 Høygradig malign | Osteosarkom, konvensjonelt Telangiektatisk osteosarkom Småcellet osteosarkom Sekundært osteosarkom Høygradig overflate osteosarkom Mesenchymalt kondrosarkom Udifferensiert høygradig malignt pleomorft sarkom (UPS) Ewing sarkom Dedifferensiert kondrosarkom Dedifferensiert kordom Lite differensiert kordom Angiosarkom |
| |||
| Variabel gradering | Konvensjonelt kondrosarkom, grad 1-3 Leiomyosarkom i ben, grad 1-3 Lav og høygradig malign kjempecelletumor i ben |
| |||
Anbefalinger:
- Ved histologisk gradering av bløtvevssarkomer anbefales det å bruke det franske graderingssystemet (FNCLCC).
- Ved histologisk gradering av sarkomer i ben anbefales en tregradig skala for konvensjonelle kondrosarkomer. Mange øvrige bensarkomer har definert grad basert på histologisk type.
Gastrointestinal stromal tumor (GIST)
GIST er den hyppigst forekommende mesenckymale tumor i gastrointestinaltraktus. Mer enn 95 % er positive for CD117 (c-kit) og DOG-1 (Fletcher et al., 2002; Miettinen & Lasota, 2006).
De fleste GIST har aktiverende mutasjoner i KIT eller PDGFRA, og typen mutasjon har betydning for prognose og valg av terapi, i tillegg til at den kan ha diagnostisk verdi. Mutasjonsanalyse skal utføres på alle GIST klassifisert som høy risiko eller ved metastaser. For den lille gruppen av GIST som ikke har mutasjoner i KIT eller PDGFRA, kan det være mutasjon i SDH (fire varianter: A, B, C og D), BRAF, KRAS eller NF-1.
Det skal avgis en risikovurdering av GIST, men tumor skal ikke malignitetsgraderes. Klassifikasjonssystemer for risikovurdering tar hensyn til tumorstørrelse, antall mitoser og lokalisasjon. Tumorruptur gir dårligere prognose og er derfor inkludert i de modifiserte NIH (National Institutes of Health) kriteriene (Joensuu, Hohenberger, & Corless, 2013).
Risikovurdering av GIST, modifiserte NIH-kriterier (Joensuu et al., 2013)

Risikovurdering av GIST, Miettinen og Lasota (Miettinen & Lasota, 2006)
| Kategori | Størrelse cm | Mitoser/ 5 mm2 | Ventrikkel | Tynntarm og alle andre lokalisasjoner, unntatt ventrikkel |
| 1 | ≤2 | ≤5 | 0 | 0 |
| 2 | >2 til ≤5 | ≤5 | 1,9 | 4,3 |
| 3a | >5 til ≤10 | ≤5 | 3,6 | 24 |
| 3b | >10 | ≤5 | 12 | 52 |
| 4 | ≤2 | >5 | 0 | 50 |
| 5 | >2 til ≤5 | >5 | 16 | 73 |
| 6a | >5 til ≤10 | >5 | 55 | 85 |
| 6b | >10 | >5 | 86 | 90 |
Anbefalinger:
- Ved risikovurdering av GIST skal det brukes et etablert risikoklassifiseringssystem, og de modifiserte NIH-kriteriene anbefales.
- Alle GIST med høy risiko og metastatisk GIST skal analyseres for mutasjoner i KIT og PDGFRA.
Rhabdomyosarkom og molekylærpatologi
Fler enn 85 % av rhabdomyosarkomer med alveolær histologi har en translokasjon av typen PAX3-FOXO1 (70 %) eller PAX7-FOXO1 (30 %), men mer sjeldne genvarianter, som blant annet ligner på det som påvises i embryonalt rhabdomyosarkom, forekommer også. Dette har prognostisk betydning. Fusjonsstatus vil nå bli brukt i valg av behandling av disse pasienter. PAX7-FOXO1 er assosiert med bedre prognose enn PAX3-FOXO1.
Anbefaling:
- For rhabdomyosarkom anbefales det at molekylærpatologisk analyse eller FISH utføres for å identifisere om PAX3-FOXO1 eller PAX7-FOXO1 translokasjon kan påvises.
Kvalitetssikring innen diagnostikk
Det er viktig at de ulike leddene og den samlede diagnose kvalitetssikres i sarkomutredning. For patologen gjelder det å ha gode interne rutiner dokumentert i det interne kvalitetssikringssystemet, samt at avdelingen er tilknyttet ekstern kvalitetssikring.
Immunhistokjemi
For immunhistokjemiske analyser bør avdelingen delta i et eksternt kvalitetssikringsprogram. Dette kan skje ved for eksempel å delta i United Kingdom National External Quality Assessment Service (UK NEQAS) eller Nordic Immunohistochemical Quality Control (NordiQC) program.
UK NEQAS er stasjonert i Sheffield i England. http://www.ukneqas.org.uk
NordiQC Nordic immunohistochemical Quality Control er stasjonert i Aalborg, Danmark; http://www.nordiqc.org/modules.php
NordiQC analyser 3 runder/år med 5–6 immunmarkører per runde.
Flowcytometri
UK NEQAS tilbyr kvalitetssikring av immunofenotyping.
EuroFlow’s tilbyr også kvalitetsprogram. http://www.euroflow.org
Molekylær patologi
UK NEQAS, MODHEM, EuroClonality, Euro MRD og Equalis tilbyr kvalitetssikringsprogram av molekylærpatologiske analyser.
Integrert diagnose
Ringtest er en metode for å sikre den avgitte diagnosen. En ringtest mellom de avdelinger som driver sarkomdiagnostikk der man systematisk går gjennom avgitte diagnoser er en metode som kan brukes. Da sarkom er en sjelden kreftform kan den gjerne etableres nasjonalt, men ideelt sett internasjonalt, for eksempel på nordisk basis.
Anbefaling:
- Ekstern kvalitetssikring bør brukes innen sarkompatologi.
Kirurgisk behandling av lokalisert sykdom
Sist faglig oppdatert: 28.02.2022
Hovedbehandlingen for maligne ben- og bløtvevssvulster er kirurgisk fjerning av svulsten med adekvate marginer. Kirurgisk behandling av sarkomer hos barn følger de samme prinsipper som for voksne, med enkelte modifikasjoner. Det vises til «Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn» (Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn: nasjonal faglig retningslinje, 2020).
Kirurgiske marginer
Sist faglig oppdatert: 28.02.2022
Kirurgisk margin er et uttrykk for hvor fullstendig man har fjernet svulsten. Kirurgisk margin vurderes på MDT-møte når histopatologisk analyse av operasjonspreparat foreligger.
Marginer registreres etter R-systemet (Wittekind, Compton, Greene, & Sobin, 2002).
R0: Makroskopisk og mikroskopisk negativ margin (ingen resttumor)
R1: Makroskopisk negativ, men mikroskopisk positiv margin (usikker resttumor)
R2: Makroskopisk gjenværende tumor
Rx: Kirurgisk margin kan ikke angis
Lokal R-status registreres uavhengig av om det foreligger fjernmetastaser.
Ved reeksisjoner angis kirurgisk margin til viabel tumor i preparatet. Hvis det ikke påvises viabel resttumor i preparatet, oppgis marginene som R0.
For abdominale og gynekologiske sarkomer angis reseksjonsmargin i utgangsorganet, ikke forholdet til den peritoneale bekledning av tumor. For retroperitoneale sarkom angis forholdet til kirurgiske reseksjonsflater. I tillegg bør det opplyses om det har vært peroperativ tumorsøl (gjelder spesielt ved abdominale sarkomer), og om svulsten var fjernet stykkevis.
Marginen klassifiseres i henhold til den minste margin som er oppnådd, dvs. det området på preparatet som har den dårligste dekning over tumor (kvantitativt og kvalitativt). Minste margin i millimeter er minste avstand målt på fiksert preparat fra tumor til tusjet overflate med kvalitativt dårligst margin. Patologen angir type vev i dette området (for eksempel fett eller bindevev/fascie) og tykkelsen på vevet som dekker tumor.
Patologen avgjør om marginen er negativ (tumorfri) og rapporterer minste avstand i fett, muskel eller løst bindevev som ikke er dekket av fascie.
Ved negativ makroskopisk og mikroskopisk margin (R0), kan kirurgisk margin spesifiseres som vid eller marginal i henhold til anbefalinger gitt av Skandinavisk Sarkomgruppe (SSG VII:4)
Marginal margin: Den minste marginen er utenfor, men nær tumor i ett eller flere områder. Mikroskopisk er marginen negativ overalt, men tumorceller kan finnes bare millimeter fra marginen.
Vid margin: Bestemmelsen av vid margin baserer seg både på inntrykket fra operasjonen og den histopatologiske rapport. Intakt fascie uten tumorinnvekst er tilstrekkelig for klassifikasjon som vid margin, uavhengig av avstanden mellom tumor og fascie. Myektomi der ubrudt fascie omgir tumor diagnostisert ved finnålsbiopsi, trenger ingen mål og klassifiseres alltid som vid margin. Det er en makroskopisk kappe av vev rundt tumor. En kappe av fett eller muskel eller løst bindevev (ikke fascie) må være minimum 10 mm tykk, målt på formalinfiksert preparat, for å kunne registreres som vid margin der fascie ikke finnes i kappen (College of American Pathologists, 2012).
Risikoen for lokalt tilbakefall er minst ved vid margin, litt høyere for marginal margin, og størst ved intralesjonell margin.
Bløtvevssarkom i ekstremiteter og trunkus
Sist faglig oppdatert: 28.02.2022
Primærtumor bør fjernes kirurgisk uten at den åpnes og omgivelsene kontamineres (Enneking, Spanier, & Goodman, 1980). Planleggingen skal baseres på klinisk og radiologisk utredning. Ingen absolutte retningslinjer for type kirurgi, dens omfang eller for type rekonstruksjon kan gis. Det anbefales liberal bruk av rotasjonslapper eller andre typer vaskulære lapper. Dette er spesielt viktig når det planlegges adjuvant strålebehandling (A. M. Davis et al., 2005; C. H. Gerrand et al., 2003; O'Sullivan et al., 1999).
Dersom et ekstremitetslokalisert bløtvevssarkom vurderes som inoperabel, og det er indikasjon for amputasjon, bør en vurdere preoperativ ILP, kjemoterapi eller strålebehandling. I noen tilfeller kan tumor da skrumpe og tillate ekstremitetsbevarende kirurgi (Eggermont, de Wilt, & ten Hagen, 2003; H. G. Smith et al., 2015).
Etter inngrepet bestemmes den kirurgiske margin av kirurgen, men veiledet av patologens makro- og mikroskopiske undersøkelse som angitt i Kirurgiske marginer.
Abdominalt og retroperitonealt sarkom
Sist faglig oppdatert: 28.02.2022
Den primære – og som regel eneste – behandling for pasienter med retroperitonealt sarkom er kirurgi. Komplett eksisjon (R0/R1) er en forutsetning for helbredelse (Gyorki & Brennan, 2014; Heslin et al., 1997; Lewis et al., 1998; Stoeckle et al., 2001), og langtidsoverlevelsen vil da være 50-60 % (Bonvalot et al., 2009; Gronchi et al., 2016; Toulmonde et al., 2014). På grunn av svulstenes størrelse og nærhet til vitale strukturer er mikroskopisk frie marginer ofte vanskelig å oppnå, og nøyaktig undersøkelse av preparatet med tanke på marginer er også vanskelig. Det vanligste kriteriet for inoperabilitet er innvekst i sentrale mesenteriale kar og aorta. Vener på bakre bukvegg vil ofte kunne reseceres/rekonstrueres. Ved mistanke om infiltrasjon i tilliggende organer, skal disse fjernes en bloc hvis mulig. Også tilliggende organer uten sikker innvekst fjernes ofte hvis det kan gjøres uten for store sekveler (colon, milt, cauda pancreatis, binyre, musculus psoas) (Bonvalot et al., 2009; Gronchi et al., 2009). Nyren vil ofte måtte fjernes, enten pga. mistenkt innvekst eller fordi ureter ikke kan spares.
Prognosen står og faller med kvaliteten av første operasjon (Gyorki & Brennan, 2014). De fleste pasienter med abdominalt residiv vil dø av sin sykdom, og nytten av residivkirurgi er usikker (Gronchi et al., 2015). Momenter som trekker i retning av residivkirurgi vil være langt residivfritt intervall, unifokal sykdom og langsom vekst (Gronchi et al., 2015; Gyorki & Brennan, 2014). Indikasjonsstilling og planlegging er en spesialistoppgave. Ved manglende symptomer vil man ofte observere situasjonen med mindre residivet truer vitale strukturer eller vil kunne bli inoperabelt ved vekst. Ofte vil man også legge inn en observasjonsperiode for å se sykdommens naturlige forløp an (et unifokalt peritonealt residiv kan vise seg å være multifokalt). Rent unntaksvis kan det være aktuelt å gjøre en inkomplett reseksjon av et residiv, f.eks. av et dedifferensiert fokus ved et ellers høyt differensiert liposarkom-residiv (Canter et al., 2008).
Abdominale sarkomer opereres som retroperitoneale sarkomer idet man tilstreber makroskopisk og mikroskopisk frie marginer uten tumorruptur. Ved GIST er det riktignok dokumentert at R1-reseksjon ikke forringer prognosen, men ruptur øker risikoen for residiv betydelig (Gronchi, Bonvalot, et al., 2020; Hølmebakk, Bjerkehagen, Hompland, Stoldt, & Boye, 2019; Hølmebakk et al., 2018). Så sant disse prinsippene etterleves, er den kirurgiske tilgangen underordnet (åpen, laparoskopisk, robotassistert kirurgi). GIST og andre abdominale sarkom metastaserer så å si aldri til lymfeknuter, og det skal derfor ikke gjøres lymfeknutedisseksjon. Dette gjør organsparende kirurgi mulig. I ventrikkel vil selv store GIST som regel kunne ekstirperes lokalt med knappe, men fri marginer, og formelle reseksjoner (distal, proksimal, subtotal, total gastrektomi) er sjelden nødvendig.
Gynekologisk sarkom
Sist faglig oppdatert: 28.02.2022
Korrekt utført kirurgi er den viktigste behandlingen for gynekologiske sarkomer, og innebærer ofte samarbeid mellom gynekolog og kirurg med kompetanse i abdominal sarkomkirurgi. De samme prinsipper som for sarkomkirurgi for øvrig bør følges, og gjentas ikke i dette avsnittet.
Total hysterektomi er vanligvis indisert ved mistanke om sarkom i uterus. Ved endometriestromasarkom (ESS) foretrekkes radikal hysterektomi der noe av parametriet tas med; dette fordi disse tumorer har betydelig tendens til innvekst i parametriet, eventuelt bare som intravaskulær invasjon, hvilket er vanskelig å vurdere preoperativt. Ved sarkom som involverer endometriet/uterinkaviteten foretas radikal hysterektomi med frilegging av øverste del av vagina. Det påsettes TEA for avsetting av en staplerrekke som lukker vagina i nivået under cervix. Deretter skylles vagina med sterilt vann. Det settes en ny staplerrad med påfølgende deling av vagina mellom de to radene. Dette for å forhindre intraabdominal spredning av tumorceller i vagina/uterus. Ved gjennomvekst av uterus må all tumor fjernes en bloc uten å forårsake ekstra søl. Ved enhver mistanke om sarkom må laparoskopisk fjernelse unngås da dette kan medføre utsæd av tumorceller i bekken/bukhule og risiko for implantasjonsmetastaser.
Det foretas ikke diagnostisk glandelstaging ved operasjon for sarkom. Sarkomer metastaserer sjelden til lymfeknuter i tidlig stadium. Ved operasjon for sarkom i avansert stadium fjernes bare lymfeknuter med sykdom.
Det foreligger ingen dokumentasjon på negativ effekt på overlevelsen av å bevare ovariene hos premenopausale kvinner med leiomyosarkomer. Residiver forekommer sjeldent, og kan behandles med en aromatasehemmer hos de som har en østrogenpåvirkelig tumor.
ESS og adenosarkom er hormonsensitive svulster og ved ESS er det observasjoner som tyder på en negativ effekt på overlevelsen hos kvinner med bevarte ovarier. Det anbefales derfor fjernelse av begge adneks hos disse kvinnene. Vi aksepterer dog bevarelse av ovariene hos premenopausale kvinner der residiver er sjeldne og behandling med aromatasehemmer kan være aktuelt hos de som har en østrogenpåvirkelig tumor.
Sarkom i mamma
Sist faglig oppdatert: 28.02.2022
Behandlingen av alle undergrupper av sarkom i mamma er kirurgisk eksisjon med mikroskopisk fri margin (R0). Prognosen er ikke bedre etter mastektomi enn lokal eksisjon, så sant fri margin oppnås, men tumors størrelse tilsier ofte mastektomi. Sykdommen sprer seg sjelden lymfatisk, og aksillær lymfeknutedisseksjon utføres ikke.
På grunn av den diffuse utbredelsen av sekundært angiosarkom, kan frie marginer være vanskelig å oppnå, og tidligere radioterapi gjør marginene vanskelige å vurdere mikroskopisk. Omfattende reseksjoner er ofte nødvendig, og rekonstruksjon for huddekning kan komme på tale. Det finnes sentra som anbefaler rutinemessig reseksjon av all bestrålt hud ved stråleassosiert angiosarkom (Feinberg et al., 2018; Morgan et al., 2012). Dette er mutilerende/kosmetisk svært skjemmende inngrep og praktiseres ikke i Norge.
Bensarkom
Sist faglig oppdatert: 28.02.2022
Kirurgi er standard behandling for de fleste pasienter med lokalisert bensarkom.
Valg av type kirurgi er avhengig av lokalisasjon, histologisk type og malignitetsgrad. Planleggingen av kirurgi bygger også på nøyaktig utførte radiologiske og nukleærmedisinske undersøkelser for å kartlegge svulstens utbredelse i skjelettet, eventuelle metastaser, utbredelse av ekstraskeletal bløtdelskomponent, og svulstenes relasjon til nerver, blodårer eller andre organer. I de aller fleste tilfeller er ekstremitetsbevarende kirurgi mulig (Zaikova et al., 2015).
Kirurgi er den eneste behandlingsmodalitet for de fleste lavgradig maligne bensarkomer og de fleste kondrosarkomer (uansett malignitetsgrad), fordi kjemoterapi og strålebehandling ikke synes å ha tilstrekkelig effekt på disse svulstene. Unntaket er svulster med vanskelig lokalisasjon som gjør adekvat kirurgi umulig, og i slike tilfeller kan definitiv stråleterapi vurderes.
Kirurgi for høygradige maligne bensarkomer er som regel en del av multimodal behandling.
De fleste bensarkomer skal fjernes med fri margin. Unntaket er kondrosarkom med malignitetsgrad 1 (atypisk kartilagenøs tumor (ACT) etter ny nomenklatur) i ekstremiteter som kan fjernes med intralesjonell utskrapning av tumormasser og eventuelt forsterkning av knokkelen med sement eller bengraft, med eller uten osteosyntese.
Ved svulster i bekken er det utfordrende å oppnå fri margin pga. komplisert anatomi. Bruk av peroperativ komputernavigasjon er vist å forbedre kirurgiske marginer og redusere antall intralesjonelle inngrep (Bosma, Cleven, & Dijkstra, 2019; Evrard, Schubert, Paul, & Docquier, 2019; Gerbers, Stevens, Ploegmakers, Bulstra, & Jutte, 2014). Peroperative hjelpemidler som komputernavigasjon eller individuelle 3D-printede reseseksjonsmaler anbefales ved operasjoner på bekkenskjelett.
Rekonstruksjoner ved ekstremitetsbevarende kirurgi
Ved ekstremitetsbevarende kirurgi for både maligne og benigne bensvulster i ekstremiteter eller bekkenskjelett blir ofte større partier av ben eller hele knokkel fjernet. Funksjonelt resultat er avhengig av både rekonstruksjonsmetode og av hvilke andre anatomiske strukturer (muskler, nerver eller blodårer) som blir fjernet.
De vanligste rekonstruksjonsmetodene er:
- Ingen rekonstruksjon. Ved fjerning av fibula, en av underarmsknoklene, clavicula, scapula, deler av bekken eller deler av ryggvirvler, trenger man ikke noen form for skjelettrekonstruksjon.
- Allograft. Man kan bruke skjelettdeler fra benbank (allograft). For rekonstruksjon av knokkel brukes det strukturert allograft. Det finnes to typer strukturerte allografter: interkallære (inneholder kun strukturert del av knokkel uten ledd) eller osteoartikulære (inneholder strukturert del av knokkel med leddflate). Rekonstruksjoner med allograft har vært mye brukt før.
- Fordeler med allograft: Alle knokler er tilgjengelige, det er lite immunologiske reaksjoner, allograft kan tilhele til pasientens eget ben, og sener og muskler kan tilhele til allograft.
- Negative sider ved allograft: Det er høy infeksjonsrisiko, innheling av allograft tar lang tid, spesielt i kombinasjon med kjemoterapi eller strålebehandling. 50 % av pasienter som er operert med allograft må reopereres pga. infeksjon eller mekaniske problemer i løpet av 10 år. Det er ressurskrevende å drive benbank. I Norge bestilles strukturerte allografter fra utlandet. Det tar tid før allograft kommer, og man risikerer at det man får ikke passer. Bruk av allograft er generelt avtagende.
- Artrodese (avstivning av ledd). Artrodese er en varig løsning som tåler belastning og er smertefri. Ulempen med artrodese er tap av bevegelighet og forkortning av ekstremitet. Ofte må man kombinere artrodese med andre rekonstruksjoner (allograft eller fibulagraft) for å bevare ekstremitetslengde. Kjemoterapi forsinker tilheling. Artrodeser brukes nå fortrinnsvis i ankelledd og i håndledd, og bare unntaksvis i andre lokalisasjoner.
- Autograft. Man kan bruke deler av knokler fra pasienten selv for rekonstruksjon. Det finnes to typer av strukturelle autograft: vaskularisert og ikke-vaskularisert. Metoden har begrenset bruksområdet og er ressurskrevende. Donorstedmorbiditet er beskrevet.
- Ekstrakorporalt behandlet autograft. Fjernet knokkel med tumor kan gjennomgå ekstrakorporal behandling for å drepe alle celler, og kan etter det reimplanteres i pasienten. Man kan bruke ioniserende stråling eller dypfrysing for ekstrakorporal behandling av autograft. De onkologiske resultatene med denne type rekonstruksjon er like gode som ved andre rekonstruksjoner, Metoden egner seg best for rekonstruksjon ved diafysær tumorlokalisasjon (Higuchi et al., 2017; Igarashi et al., 2014).
- Tumorproteser. Dette er mest brukt rekonstruksjonsmetode. Alle lange rørknokler og store ledd kan rekonstrueres. Moderne modulære tumorproteser gir stor fleksibilitet og muligheter for individuell tilpasning. I noen spesielle tilfeller kan man benytte spesiallagede proteser (custom made). Kjente komplikasjoner ved tumorproteser er infeksjon, mekanisk løsning, brekkasje og slitasje av komponenter. Tumorpasienter er ofte unge mennesker som vil leve aktivt og bruke sine proteser i mange tiår. Økt bruk av tumorproteser vil gi økt behov for revisjonskirurgi. Infeksjonsrisiko ved bruk av tumorproteser ligger på ca 15 %, rapportert fra flere tumorsentra internasjonalt. I de siste årene har det kommet rapporter om at bruk av tumorproteser med sølvbelegg reduserer infeksjonsrisiko, selv om denne kunnskapen er bygget på ikke-randomisert studie (evidensnivå C), (Hardes et al., 2010; Schmidt-Braekling et al., 2017; Wafa et al., 2015).
- Skjelettrekonstruksjoner hos barn. Voksende skjelett gir utfordringer for rekonstruksjonsmetoder. Så langt som mulig forsøker en å bevare vekstsonene (epifyseskivene), og anvende biologisk skjelettrekonstruksjon. Allikevel opereres de fleste barn med bensarkom med tumorproteser fordi biologisk rekonstruksjon enten er umulig eller ikke forventes å gi tilfredsstillende funksjonelt resultat. Ved planlegging av kirurgi beregner man gjenværende vekst i knokkel som er berørt av tumor. Dersom man forventer utvikling av anisomeli (benlengdeforskjell) på over 3 cm kan en vurdere å bruke ekspanderbare proteser (vokseproteser). Postoperativt skal det gjøres regelmessige benlengdemålinger og behov for epifyseodese (kunstig lukking av veksteskivene) skal vurderes.
- Rotasjonsplastikk. Dersom en må fjerne hele eller deler av lårbenet hos barn i vekst kan det være aktuelt å bruke denne metoden. Ved rotasjonsplastikk monteres ankelen på kneets plass rotert 180 grader, slik at fotbladet kan styre en leggprotese, og ankelen vil fungere som et kneledd. Metoden gir høyt funksjonsnivå sammenlignet med femuramputasjon, men oppleves ofte som kosmetisk sjenerende av pasienter. Hvis rekonstruksjon med tumorprotese er mulig, blir derfor tumorprotese oftest foretrukket. Når man planlegger kirurgi med rotasjonsplastikk er det meget viktig at både barnet selv og familien får grundig informasjon om hva dette innebærer i god tid før operasjon.
- Rekonstruksjon ved bekkenreseksjon. Kirurgi for bensarkomer i bekkenregion er oftest ressurskrevende, komplisert og innebærer en vesentlig reduksjon av funksjon hos pasienten. Peroperativ bruk av computernavigering har vist å ha en positiv effekt på kirugiske marginer og bør benyttes ved kirurgi for bensvulster i bekkenregion. Rekonstruksjonsmetode er avhengig av type reseksjon. Mest utfordrende er å rekonstruere hofteledd når deler av bekken med acetabulum er fjernet.
Bensarkomer i ryggsøylen
Bensarkomer i ryggvirvler er sjeldne. Tumor kan vokse inn i spinalkanalen og forårsake nevrologisk utfall. Ofte er dette årsaken til at pasienten tar kontakt med helsevesenet. På grunn av faren for utvikling av permanent motorisk utfall, må disse pasientene henvises raskt til sarkomsenter, ofte som øyeblikkelig hjelp, og uten forutgående biopsi eller kirurgi. I enkelte tilfeller kan man forvente en god og rask effekt av kjemoterapi (for eksempel ved Ewing sarkom) og kan derfor unngå akutt intralesjonal kirurgi for å avlaste ryggmargen. Planlegging av kirurgi bør foregå i tett samarbeid mellom tumorortoped og ryggkirurg eller nevrokirurg.
Rehabilitering
Funksjon etter kirurgi for bensarkomer er avhengig av hvilke strukturer som er fjernet (muskler, ledd, nerver og blodårer) og rekonstruksjonsmetode, men også av adekvat og individuelt tilpasset opptrening etter kirurgi, både i den tidlig postoperative fasen og senere. Pasienter som er operert med tumorproteser (og andre rekonstruksjonsteknikker), bør få individuell oppfølging av fysioterapeuter med kompetanse innen denne type reseksjons- og rekonstruksjonsmetoder.
Kirurgi som en del av tverrfaglig og multimodal behandling
Sist faglig oppdatert: 28.02.2022
En betydelig andel sarkompasienter med lokalisert sykdom får multimodal behandling. Alle slike pasienter skal vurderes og utredes i regi av et multidisiplinært team på sarkomsenter. I mange tilfeller vil det være behov for gjentatte diskusjoner på multidisiplinært møte, f.eks. i forbindelse med preoperativ evaluering og gjennomgang av operasjonspreparat. Det er ofte komplekse vurderinger, der mange hensyn skal veies mot hverandre, og der er en stor grad av individualisering er nødvendig. Det er således vanskelig å legge entydige føringer for slike vurderinger.
Sarkomer kan oppstå i alle anatomiske lokalisasjoner. Det betyr at det er flere kirurgiske spesialiteter (f.eks. ortopeder, gastrokirurger, nevrokirurger, thorakskirurger og ØNH-kirurger) som kan være involverte i behandling av sarkompasienter.
Sarkom i andre lokalisasjoner
Sist faglig oppdatert: 28.02.2022
Sarkom kan oppstå i enhver anatomisk lokalisasjon. Kirurgisk behandlingen av sarkomer i andre lokalisasjoner vil ikke bli omtalt spesielt, men skal som hovedregel følge de samme prinsipper som for de lokalisasjoner som er omtalt. I enkelte lokalisasjoner kan tilstrekkelig margin være vanskelig å oppnå, f.eks. i hode/hals, og terskelen bør i slike situasjoner være lav for multidisiplinær diskusjon på tvers av helseregioner.
Anbefalinger:
- Kirurgi er standard behandling for de fleste lokaliserte bløtvevssarkomer og bensarkomer.
- Ekstremitetsbevarende kirurgi anbefales som hovedregel ved ekstremitetslokaliserte sarkomer.
- Ved abdominale, retroperitoneale og gynekologiske sarkomer tilstrebes operasjon med makroskopisk og mikroskopisk frie marginer uten tumorruptur.
- Preoperativ planlegging og utredning av pasienter med sarkom skal foretas innen multidisiplinært team på et sarkomsenter
- Preoperativ behandling av bløtvevssarkomer vurderes individuelt på bakgrunn av tumorlokalisasjon, operabilitet og histologisk diagnose.
- Hjelpemidler som peroperativ komputernavigasjon eller individuelle 3D-printede reseseksjonsmaler anbefales ved operasjoner på bekkenskjelett.
Strålebehandling og annen lokalbehandling ved lokalisert sykdom
Generelt om strålebehandling ved sarkom
Sist faglig oppdatert: 28.02.2022
Det vises til Skandinavisk sarkomgruppes retningslinjer for strålebehandling ved ben- og bløtvevssarkom (SSG XXIV, desember 2015), publisert på https://www.ssg-org.net/, utarbeidet av en tverrfaglig skandinavisk arbeidsgruppe. Norske sarkommiljøer er en del av SSG, har bidratt aktivt til protokollen, og følger disse anbefalingene.
Spesielle betraktninger gjelder ved strålebehandling av barn, og det vises her til Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn (Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn: nasjonal faglig retningslinje, 2020).
Indikasjoner: Strålebehandling kan være indisert som preoperativ (neoadjuvant) eller postoperativ (adjuvant) behandling (Al-Absi et al., 2010; Kepka, DeLaney, Suit, & Goldberg, 2005; O'Sullivan et al., 2002; P. W. Pisters, O'Sullivan, & Maki, 2007; C. S. Trovik et al., 2001; Yang et al., 1998), (Evidensgrad A), som del av definitiv lokalbehandling med kurativ intensjon (Kepka et al., 2005), eller som palliativ behandling, både ved ben- og bløtvevssarkom.
Teknikker: Typisk benyttes fraksjonert behandling med foton- eller elektronstråling, eventuelt kombinasjoner av disse. Hos barn, og i spesielle tilfeller der tumor har nær relasjon til kritiske sentralnervøse strukturer, kan protonstråling gi mer presis dosefordeling samt bedre skjerming av normalvev. Det foreligger få kliniske studier som sammenligner ulike behandlingsteknikker ved strålebehandling av sarkom. En bekymring som er reist er om invers planlegging med IMRT (intensitetsmodulert radioterapi) eller VMAT (Volumetric Modulated Arc Therapy); teknikker som medfører utsmøring av dose og derved lavdosestråling til større volum sammenlignet med konvensjonell 3D-planlegging (3DRT), på lang sikt kan innebære økt risiko for stråleassosiert kreft. I en fersk dyrestudie (Gomarteli et al., 2019) hvor slik invers planleggingsteknikk sammenlignes med 3D-planlegging, finner man imidlertid ikke holdepunkter for at større lavdosevolumer innebærer økt risiko for sekundærkreft. En klinisk studie på ekstremitetslokalisert bløtvevssarkom konkluderer med at VMAT gir bedre sparing av normalvev sammenlignet med 3DRT (Di Brina et al., 2019). Valg av teknikk må baseres på optimalisering av balansen mellom effekt og bivirkninger (Evidensgrad D).
Fiksering og behandlingsposisjon
Siden optimal fiksering og strålebehandlingsposisjon er vanskelig å standardisere (ryggleie, bukleie, sideleie, fleksjon eller ekstensjon i ulike ledd), er det nødvendig med nært samarbeid om den enkelte pasient mellom ansvarlig onkolog, fysiker og stråleterapeut som har erfaring med strålebehandling ved ben- og bløtvevssarkom. Fiksasjon med termoplastisk materiale festet til skinner spesiallaget for ulike ekstremitetsdeler er å foretrekke med tanke på reproduserbarhet av behandlingsleie/presisjon.
Bruk av kontrastmidler og bolusmateriale
Intravenøs kontrast kan brukes når framstilling av karstrukturer anses viktig for definisjon av målvolum og kritiske organer, og gir i tillegg bedre kontrast mellom ulike typer bløtvev. Blystreng på arr kan lette fremstillingen på CT-matrisen dersom arrområdet skal inngå i målvolumdefinisjonene.
Når utvendig bolusmateriale er nødvendig for å oppnå optimal dosefordeling, kan det være hensiktsmessig å legge på individualisert bolus allerede ved CT-opptaket, spesielt hvis arret kommer i direkte kontakt med fiksasjonsutstyr eller behandlingsbord. Bolus for å sikre dekning av arr med f.eks. 2 cm margin i målvolumet er indisert ved intralesjonell margin. Bolus anbefales også etter marginal margin i tilfeller hvor tumor diffust infiltrerer hud eller underhud som ikke er fjernet en bloc med tumor, og kan vurderes etter primær reseksjon eller insisjonsbiopsi utført utenfor sarkomsenter. Ved bruk av bolus vil den tilsiktede økte huddosen ofte medføre betydelig dermatitt/deskvamasjon.
Volumdefinisjoner og marginer
Dersom pasienten er inkludert i spesifikk protokoll, følges tilhørende anbefalinger om strålebehandlingsteknikk.
Klinisk målvolum (Clinical Target Volume, CTV) og doseplanleggingsvolum (Planning Target Volume, PTV) defineres for hver pasient. Ved preoperativ strålebehandling og definitiv strålebehandling tas utgangspunkt i makroskopisk tumorvolum (Gross Tumour Volume, GTV). Dersom målvolumet er vanskelig å avgrense ved CT, kan en fusjonere diagnostiske bildesekvenser, for eksempel CT, MR og eventuelt PET/CT med CT-matrisen (Wang et al., 2011). Ved inkomplett anatomisk sammenfall mellom bildemodalitetene, kan det være hensiktsmessig med summasjon av GTV definert ved forskjellige bildemodaliteter med Boolean teknikk (Daisne et al., 2004).
Ved postoperativ strålebehandling kan det være vanskelig å definere operasjonsvolumet («sårsengen») som utgangspunkt for å bestemme marginer til CTV. Et opprinnelig GTV kan gjenskapes ved fusjonering av preoperativ radiologisk diagnostikk, samt ved koregistering av postoperative MR-sekvenser som viser økt vannholdighet/ødem, f.eks. STIR-sekvenser. Ved postoperativ strålebehandling er det nødvendig med nært samarbeid med opererende kirurg for definisjon av målvolum.
Fra GTV til CTV har et internasjonalt ekspertpanel anbefalt en margin på 4 cm i longitudinell retning, og 1,5 cm radialt (Dickie, Haas, & O'Sullivan, 2015; Haas et al., 2012). CTV kan formes med knappere marginer mot anatomiske barrierer som f.eks. periost og muskelfascier som ikke radiologisk er engasjert av tumor, eller mot kritiske organer. CTV tilpasses for øvrig utbredelsen av operasjonsområdet, drenskanal, samt postoperative forandringer som serom/lymfocele/hematom.
Marginene fra CTV til PTV må individualiseres avhengig av anatomisk lokalisasjon og institusjonens egen erfaring ved aktuelle organlokalisasjon. En isotropisk margin på 0,5–1,0 cm i alle retninger fra CTV er veiledende, med eventuelle justeringer relatert til immobilisering, reproduserbarhet og tilgjengelig billeddiagnostikk for feltkontroller (Haas et al., 2012). Ved behandling av ekstremiteter er det ofte nødvendig med større marginer enn i f.eks. hode/hals-området. Ved regelmessig (fortrinnsvis daglig) bruk av radiologisk avbildning på strålebehandlingsbordet, enten med kilovoltteknikk («on board imaging») eller «cone beam CT», kan reproduserbarheten mellom behandlingsfraksjonene økes og marginene mellom CTV og PTV reduseres.
Kritiske organer og dosering
Definisjon av kritiske organer er avhengig av involvert anatomisk område og planlagt dosenivå, og er vanskelig å standardisere. Det anbefales liberal definisjon av mulige kritiske organer, hvilket også gjør rapportering av dose-volumparametre til nærliggende organer lettere.
Ved tilleggsdose til høyrisikovolum ved intralesjonell margin og ved definitiv strålebehandling, kan det oppstå dosebegrensende toksisitet i kritiske organer. Ved valg av dose til PTV må en ta hensyn til estimert risiko for organtoksisitet basert på publiserte empiriske data (Emami et al., 1991; Marks et al., 2010; Milano, Constine, & Okunieff, 2007). (Evidensgrad B).
Valg av strålebehandlingsplan
Ved valg av behandlingsplan tas det hensyn til grad av konformalitet av dosefordeling i PTV samt dose til kritiske organer. En kan bruke den strålebehandlingsplanen som gir mest optimal dosefordeling, enten ved hjelp av konvensjonell 3D konformal teknikk, intensitetsmodulert strålebehandling (IMRT), buestrålebehandling (VMAT) eller partikkelstråling (Alektiar, Brennan, Healey, & Singer, 2008).
Forskriving, lagring av data og rapportering av strålebehandling
Dette følger retningslinjene fra International Commission on Radiation Units and Measurements (ICRU 50, 62 og 85a etc). Direktoratet for strålevern og atomsikkerhet (www.nrpa.no) har i Strålevernrapport 2012: 9 «Volum og doser i ekstern stråleterapi», beskrevet definisjoner for volum- og doseparametre, samt anbefalinger for bruk, dokumentasjon og rapportering av strålebehandling (Levernes & Johannessen, 2003).
Strålebehandling ved bensarkom
Sist faglig oppdatert: 01.02.2022
De aller fleste pasientene med Ewing sarkom eller osteosarkom behandles etter internasjonale behandlingsprogrammer. Slike protokoller har ofte detaljerte retningslinjer for strålebehandling som definerer beste internasjonale behandlingsstandard.
For Ewing sarkom har dels protokollene ISG/SSG III og IV vært benyttet som retningslinje for voksne, dels Euro Ewing 2012. Sistnevnte har vært foretrukket av de pediatriske miljøene i Skandinavia (S. Ferrari et al., 2011; Luksch et al., 2012). Det anbefales at indikasjon for strålebehandling og fraksjonering følger de ovennevnte protokollene. Lokal kontroll etter definitiv strålebehandling ved Ewing sarkom er relativt god, og bør vurderes som alternativ til kirurgi dersom kirurgi er mutilerende. Det foreligger ingen randomiserte data der definitiv strålebehandling og kirurgi er sammenlignet.
Osteosarkom er en lite strålefølsom tumor, og definitiv strålebehandling gir lavere sannsynlighet for lokal kontroll enn kirurgi. Dersom kirurgi ikke er mulig vil likevel definitiv strålebehandling være anbefalt. Totaldose ≥70 Gy er anbefalt.
Ved kondrosarkom er det sjelden indikasjon for strålebehandling, men det kan overveies ved marginal og intralesjonell margin ved høygradig kondrosarkom, ved inoperabilitet, utfordrende anatomisk lokalisasjon som f.eks. skallebasis, eller ved residiv. Kondrosarkom anses som en relativt lite strålefølsom subtype (Riedel et al., 2009).
Ved kordom i sakrum, coccyx, columna eller skallebasis/os sphenoidalis, kan strålebehandling være indisert ved intralesjonell margin eller som definitiv behandling (Jian et al., 2010). Da bør protonbestråling/tyngre ioner også overveies (Demizu et al., 2017; Uhl et al., 2014) (Evidensgrad C).
Strålebehandling ved bløtvevssarkom
Sist faglig oppdatert: 28.02.2022
Strålebehandling ved bløtvevssarkom i ekstremiteter og trunkus
Det vises til skandinaviske retningslinjer (ssg-org.net). Skandinaviske data har bidratt til økt evidens for nytten av strålebehandling for lokal kontroll ved bløtvevssarkom (N. L. Jebsen et al., 2011; N.L. Jebsen et al., 2008). Dette innbærer indikasjon for postoperativ strålebehandling ikke bare ved marginal og intralesjonell margin, men også ved vid margin ved høygradig maligne dype svulster, samt i kliniske situasjoner der eventuelt residiv medfører høy sannsynlighet for mutilerende kirurgisk behandling (se tabell nedenfor). Nytten av av strålebehandling som adjuvans til kirurgi ved lokalisert bløtvevssarkom i ekstremiteter og kroppsvegg er nylig bekreftet i en metaanalyse (Albertsmeier et al., 2018; Gannon et al., 2019).
Der er en økende trend internasjonalt mot å anvende preoperativ strålebehandling ved høygradige bløtvevssarkom fordi målvolumet ofte reduseres og definisjon av målvolumet forenkles. Stråledose til normalvev begrenses dermed, hvilket innebærer at seneffekter i normalvev slik som fibrose, leddstivhet og ødem synes å kunne reduseres. Preoperativ strålebehandling gir imidlertid økt risiko for sårkomplikasjoner og redusert funksjonsnivå i tilhelingsfasen 1. Relatert til høyere risiko for seneffekter ved større målvolum er forekomst av seneffekter rapportert å være høyere og funksjonsnivå lavere etter postoperativ strålebehandling sammenlignet med preoperativ tilnærming (A. M. Davis et al., 2005). Lokal kontroll er evidensmessig like god med preoperativ og postoperativ behandling.
Den skandinaviske adjuvante protokollen SSG XX for voksne pasienter med lokalisert høyrisiko bløtvevssarkom innebærer bruk av akselerert hyperfraksjonert strålebehandling i kombinasjon med kirurgisk behandling og adjuvant kjemoterapi med doksorubicin og ifosfamid. Med oppfølgingstid på median 3,9 år (150 inkluderte pasienter hvorav 131 fikk postoperativ strålebehandling) var lokal kontroll etter 5 år i Arm A 14 % (Sundby Hall et al., 2018). SSG XX Arm B inkluderte lokalavanserte, høygradige tumores hvor risiko for intralesjonell kirurgi var vurdert som betydelig. Disse pasientene gjennomgikk akselerert strålebehandling pluss kjemoterapi før kirurgi, med tillegg av kjemoterapi postoperativt (Hall et al., 2020). Få tilfeller av lokalt residiv (3/20) i denne ekstra høyrisikogruppen motiverer for en preoperativ tilnærming ved vanskelig resekterbare tumores. Med bakgrunn i gode overlevelsesdata (5-års totaloverlevelse på 76.1%), er behandlingsregimet fra SSG XX etablert som standard ved høyrisiko bløtvevssarkom i ekstremiteter og trunkus (Evidensgrad B).
Strålebehandling kan også være indisert som definitiv lokal terapi ved inoperabilitet, eller der pasienten ikke ønsker eller tåler å gjennomgå kirurgisk behandling.
Effekten av bestråling av lavgradige sarkom har vært diskutabel. Jebsen et al. (N.L. Jebsen et al., 2008) viste imidlertid høyere lokal kontrollrate etter strålebehandling på 93 % i gruppen subkutane lavgradige bløtvevvsarkom operert med intralesjonell margin, sammenlignet med 82 % i gruppen behandlet med kirurgi alene. For dype lavgradige, intralesjonelt opererte var forskjellen 90 % versus 75 % med og uten strålebehandling. Dataene bekrefter statistisk signifikant nytte av strålebehandling også ved lavgradige bløtvevssarkom. Den kliniske nytteverdien må vurderes individuelt i multidisiplinære team, og baseres blant annet på i hvor stor grad et lokalt residiv kan fjernes uten mutilerende kirurgi.
Det er internasjonalt begrenset enighet om optimalt dosenivå ved strålebehandling ved primært bløtvevssarkom. I Skandinavia har anbefalingen vært 50 Gy/25 fraksjoner etter vid margin ved dyptliggende svulster og ved marginal margin. Andre organisasjoner anbefaler høyere dose. Ved intralesjonell margin er det aktuelt med 10–20 Gy som tilleggsdose til et definert høyrisikovolum (Alektiar et al., 2000; Wolfson et al., 1998; Zagars & Ballo, 2003). Ved definitiv strålebehandling kan enda høyere dosenivå være ønskelig dersom det er mulig med høy dose til et begrenset volum, og dersom dosene til kritiske organer ansees forsvarlige. Det er holdepunkter for at 68 Gy eller høyere øker sannsynligheten for lokal kontroll (Fein et al., 1995; Zagars & Ballo, 2003) (Evidensgrad B).
Myxoide liposarkom er vist å være spesielt strålefølsomme (Engström et al., 2007). Norge har deltatt i en studie med redusert stråledose i neoadjuvant setting (DOREMY) i form av 36 Gy/18 fraksjoner mot standard 50Gy/25 fraksjoner. De nylig publiserte resultatene viste ypperlig lokal kontroll og lavere forekomst av senskader etter strålebehandling sammenlignet med historiske data (Lansu et al., 2021). Dersom preoperativ strålebehandling er indisert ved myxoid liposarkom er det konsensus i Norge for å benytte redusert stråledose til 36 Gy. For å styrke evidensgrunnlaget for optimalisering av strålebehandling ved denne subtypen, deltar Norge i et nyopprettet internasjonalt, prospektivt register for lokal behandling av myxoid liposarkom.
%20ved%20bl%C3%B8tvevssarkom.png)
Strålebehandling ved abdominalt og retroperitonealt sarkom
Det ble i 2008 aktivert et skandinaviske handlingsprogram for intraabdominale, retroperitonale og pelvine sarkom, SSG XVII (www.ssg-org.net). Basert på retrospektive skandinaviske data, samt internasjonale prospektive data, åpnet dette programmet for strålebehandling ved intralesjonell margin etter reseksjon av høygradige svulster der man vurderte lokal tilleggsbehandling som hensiktsmessig (M. J. Smith et al., 2014; L. H. Trovik et al., 2014). Resultater fra STRASS-studien gir imidlertid ikke støtte for rutinemessig bruke av strålebehandling ved retroperitoneale sarkom (RPS) (Bonvalot et al., 2020). Studien randomiserte mellom preoperativ strålebehandling før en bloc kirurgi eller kirurgi alene. Pasienter med resektabel, unifokal, ikke-metastatisk sykdom beliggende retroperitonealt eller infraperitonealt i bekkenet med histologisk bekreftet RPS ble inkludert. Resultater fra denne studien viste ingen signifikant bedret lokal residivfri overlevelse i gruppen som fikk strålebehandling (van Houdt et al., 2019). Lignende utfall er rapportert i en retroprospektiv studie fra US Sarcoma Collaborative (Chouliaras et al., 2019), og det foreligger således ikke evidens for generell nytte av preoperativ strålebehandling ved retroperitonealt sarkom. Imidlertid indikerte en post hoc subanalyse av pasienter med liposarkom og lavgradige RPS bedre overlevelse i gruppen som fikk strålebehandling før kirurgi sammenlignet med gruppen som gjennomgikk kirurgi alene. I spesielle tilfeller av vanskelig resektable liposarkom eller lavgradige sarkom hvor man forventer ufri margin, kan preoperativ strålebehandling diskuteres. Postoperativ strålebehandling ved RPS har tilsvarende begrenset evidens, og er forbundet med betydelig akutt- og sentoksisitet, men kan i følge ESMO-EURACAN guidelines appliseres i spesielle tilfeller til et veldefinert anatomisk høyrisikoområde (Gronchi et al., 2021).
Strålebehandling kan også være indisert som definitiv behandling ved ikke-resektable svulster, men grunnet dosebegrensning som skyldes omliggende kritiske strukturer må slik behandling ansees som palliativ.
Strålebehandling ved gynekologisk sarkom
Det foreligger ingen studier som har vist gevinst av adjuvant stråleterapi ved gynekologiske sarkomer. Stråleterapi kan overveies ved lokalisert tumor som ikke egner seg for kirurgi og har en lokalisasjon som er egnet for strålebehandling
Strålebehandling av barn med sarkom
De spesifikke internasjonale protokollene for barn og ungdom med sarkom inneholder egne kapitler om strålebehandling; det gjelder f.eks. EpSSG-protokollene for rhabdomyosarkom (FaR-RMS) og non-rhabdomyosarkom, samt protokoller for osteosarkom (EURAMOS 1) og Ewing sarkom (Euro Ewing 2012). FaR-RMS er åpen for inklusjon og har flere studiespørsmål relatert til strålebehandling, f.eks. pre- vs postoperativ strålebehandling, doseeskalering og bestråling av primærtumor vs alle lesjoner ved metastatisk sykdom. De andre protokollene rekrutterer ikke lenger aktivt, men brukes som retningslinjer for behandling.
Partikkelterapi ved sarkom
Sist faglig oppdatert: 28.02.2022
Der foreligger ingen randomiserte studier som sammenligner fotonstrålebehandling med partikkelbestråling hos pasienter med sarkomer. Basert på tilgjengelig evidens konkluderte en ekspertgruppe tilknyttet American Society of Radiation Oncologi i 2012 at protonterapi er fordelaktig sammenlignet med fotonstråling ved kondrosarkomer i aksiale skjelett/skallebasis og kordom, i tillegg til ved pediatriske svulster i sentralnervesystemet samt store øyemelanomer (Allen et al., 2012; Noel et al., 2003). Spesielt hos barn, der langtidstoksisitet er en betydelig utfordring, kan bruk av protoner teoretisk redusere risiko for seneffekter, inkludert stråleassosiert kreft (Miralbell, Lomax, Cella, & Schneider, 2002). Det fysiske og strålebiologiske grunnlaget for overlegen dosefordeling (dvs. sparing av normalvev) ved protoner vs. fotoner er et argument for at positive fase III studier ikke synes å være en forutsetning for å implementere partikkelterapi (Suit et al., 2008). Sarkomers sjeldenhet underbygger dette argumentet. Det foreligger likevel klinisk dokumentasjon på nytte av partikkelterapi ved sarkom (Thomas & Timmermann, 2020; Weber et al., 2017). Sarkomer i sentralakselokalisasjon og hos barn blir i økende grad behandlet med protonterapi verden over.
Protonterapi synes å være gunstig ved kordom, spesielt ved skallebasislokalisasjon og kraniale deler av sakrum, for å tillate eskalering av stråledosen og dermed øke sannsynlighet for lokal kontroll (Igaki et al., 2004; Noel et al., 2005). God lokal kontroll med begrenset behandlingsrelatert toksisitet er vist i studier som har inkludert både kordom og kondrosarkom i skallebasis eller cervikalcolumna (Habrand et al., 2008; Noel et al., 2003; Rosenberg et al., 1999). Oppmuntrende resultater er publisert også ved ikke-resektable eller inkomplett resiserte bensvulster som Ewing sarkom og osteosarkom (Ciernik et al., 2011; Rombi et al., 2012; Weber et al., 2017). Dose-distribusjonsstudier har vist at protoner gir bedre konformering og sparing av normalvev hos pediatriske pasienter med orbitalt eller parameningealt rhabdomyosarkom og pelvine sarkom, samt ved intraabdominale og paraspinale bløtvevssarkom (Hug, Adams, Fitzek, De Vries, & Munzenrider, 2000; C. T. Lee et al., 2005; Mizumoto et al., 2018; Swanson et al., 2012; Thomas & Timmermann, 2020; Weber, Trofimov, Delaney, & Bortfeld, 2004). Partikkelterapi som involverer tyngre ioner er mer eksperimentelt, men eksempelvis er karbonioner rapportert som en effektiv behandling med moderat toksisitet ved både ben- og bløtvevssarkom i ulike lokalisasjoner (Jingu et al., 2012; Kamada et al., 2002).
Rasjonalet for å velge partikkelstråler (i praksis protoner) hos pasienter med sarkom bygger på 1) mer gunstig dosefordeling og derved sparing av normalvev; samt 2) et større potensiale for kurasjon etter definitiv strålebehandling til høye doser ved relativt stråleresistente, primære bensvulster som har en beliggenhet som umuliggjør kirurgisk reseksjon. Grunnet reduserte langtidseffekter, er det internasjonal enighet om at sarkom hos barn generelt skal vurderes for partikkelstråling. Alle helseregionene i Norge har egne protongrupper med utstrakt nasjonalt samarbeid, og kontaktes om aktuelle pasienter.
Regional varmebehandling (hypertermi)
Sist faglig oppdatert: 28.02.2022
Ved Senter for ben- og bløtvevssvulster, Haukeland universitetssykehus, er det siden 1994 erfaring med regional varmebehandling (hypertermi) ved lokalavansert ikke-metastatisk bløtvevsarkom. Etter fase II-erfaring med neoadjuvant termokjemoterapi rekrutterte en pasienter med høyrisiko bløtvevssarkom til en internasjonal fase III-studie der en randomiserte mellom neoadjuvant kjemoterapi med etoposid, ifosfamid og doksorubicin versus samme kjemoterapi forsterket av regional varmebehandling. Strålebehandling ble gitt postoperativ etter gjeldende retningslinjer. Studien rekrutterte 341 pasienter, hvorav 20 fra Norge, og viste signifikant bedret både sykdomsfri overlevelse og lokal kontroll i hypertermiarmen (R. D. Issels et al., 2010). Nylig presenterte data fra langtidsoppfølging viser en totaloverlevelsesgevinst på 11 % etter 9 år (54 % vs. 43 %) sammenlignet med kjemoterapi alene (R. Issels & Lindner, 2016).
Etter at denne protokollen ble lukket, har det siden desember 2006 vært aktivert en modifisert protokoll der ikke bare termokjemoterapi med doksorubicin og ifosfamid gis neoadjuvant, men også termokjemoradioterapi med strålebehandling, konkomitant ifosfamid og regional varmebehandling – «trimodal neoadjuvant behandling».
Regional varmebehandling (hypertermi) ved lokalisert bløtvevssarkom er vurdert i NyeMetoder, og er ikke innført til behandling av høyrisiko bløtvevssarkom, jf. Beslutningsforum 24.feb. 2020;: «Regional varmebehandling innføres ikke til behandling av høyrisiko bløtvevssarkom. Det er usikkerhet vedrørende dokumentasjon for effekt og bivirkninger av metoden, Beslutningen vil kunne tas opp til ny vurdering når resultater fra nyere studier foreligger. Inntil nye resultater foreligger, skal regional varmebehandling som tillegg til etablert behandling av høyrisiko sarkom kun skje i regi av kliniske studier.»
ILP
Sist faglig oppdatert: 28.02.2022
ILP (isolated limp perfusion) med tumornekrosefaktor (TNF) og melfalan utføres ved Oslo universitetssykehus, Radiumhospitalet, og kan være effektiv preoperativ behandling for å minske størrelsen av ekstremitetslokaliserte bløtvevssarkom slik at amputasjon kan unngås. Erfaringer viser at opptil 75 % kan ha nytte av behandlingen. Metoden er ikke vurdert iNye Metoder.
Anbefalinger:
- Det anbefales at gjeldende protokoller og handlingsprogram følges, for eksempel fra Skandinavisk sarkomgruppe (www.ssg-org.net).
- Ved høygradig maligne bløtvevssarkomer i ekstremiteter og trunkus anbefales postoperativ strålebehandling uavhengig av margin ved alle dypt beliggende svulster og etter marginal og intralesjonell margin uavhengig av dybde.
- Postoperativ strålebehandling anbefales etter intralesjonell margin ved bløtvevssarkomer i ekstremiteter og trunkus, uavhengig av malignitetsgrad, og i andre kliniske situasjoner der lokalt residiv medfører høy sannsynlighet for mutilerende kirurgisk behandling.
- Preoperativ strålebehandling kan vurderes i stedet for postoperativ behandling ved bløtvevssarkomer i ekstremiteter og trunkus.
- Definitiv strålebehandling bør vurderes ved Ewing sarkom dersom kirurgi blir mutilerende, og ved andre bensarkomer dersom kirurgi ikke er mulig.
- Pre- eller postoperativ strålebehandling skal som hovedregel ikke gis ved abdominalt, retroperitonealt eller gynekologisk sarkom.
- Alle barn med sarkom, og voksne pasienter med sarkom i spesielle lokalisasjoner, skal vurderes for protonbestråling.
- Regional varmebehandling skal kun tilbys innen kliniske studier.
Medikamentell behandling ved lokalisert sykdom
Medikamentell behandling av lokalisert bløtvevssarkom
Sist faglig oppdatert: 28.02.2022
Bløtvevssarkom i ekstremitet og trunkus
Kliniske studier med adjuvant eller neoadjuvant cytostatikabehandling ved lokalisert bløtvevssarkom har vist motstridende resultater. Enkelte store studier har vist liten eller ingen gevinst av adjuvant kjemoterapi (Pervaiz et al., 2008; Woll et al., 2012), mens andre har vist en overlevelsesgevinst (Frustaci et al., 2001; Gronchi, Palmerini, et al., 2020). Subgruppeanalyser har vist at pasienter med den høyeste risikoen for tilbakefall sannsynligvis har størst nytte av adjuvant kjemoterapi (Pasquali et al., 2019). Dersom adjuvant eller neoadjuvant kjemoterapi skal gis er det enighet om at det skal være en kombinasjon av et antrasyklin og ifosfamid.
De skandinaviske anbefalingene (SSG; www.ssg-org.net) er basert på en enarmet fase II -studie (SSG XX) der pasienter med høy risiko for tilbakefall fikk seks kurer doksorubicin 60 mg/m2 og ifosfamid 6 g/m2 postoperativt (Sundby Hall et al., 2018). Estimert metastasefri overlevelse etter 5 år var 70,4 %, hvilket var bedre enn historiske kontroller. I og med at studien var enarmet er det ikke mulig å konkludere sikkert om den systemiske behandlingen bedrer overlevelse.
Høyrisikogruppen i SSG XX-studien ble valgt ut basert på histologi og malignitetsgrad (FNCLCC) 2 eller 3, samt minst ett av følgende kriterier:
- Karinvasjon.
- Minst to av følgende: Tumorstørrelse ≥8 cm, tilstedeværelse av nekrose og diffust infiltrerende vekstmønster.
Alle høygradig maligne histologiske subtyper inngikk, unntatt ekstraskeletalt osteosarkom og kondrosarkom, Ewing sarkom, rhabdomyosarkom, Kaposis sarkom, malignt mesenkymom, klarcellet sarkom, alveolært bløtdelssarkom og epitelioid sarkom.
Adjuvant eller neoadjuvant kjemoterapi ved lokalisert bløtvevssarkom oppfattes ikke som standard behandling internasjonalt fordi det ikke foreligger randomiserte studier som entydig viser en overlevelsesgevinst. Europeiske retningslinjer uttaler at det bør diskuteres med pasienter som har høy risiko for tilbakefall (Gronchi et al., 2021). Det anbefales at pasienter som oppfyller inklusjonskriteriene i SSG XX-studien informeres om, og vurderes for, adjuvant kjemoterapi (Evidensgrad B). Pasienter med høyest risiko har sannsynligvis størst nytte av behandlingen, og dette bør tas med i vurderingen. Neoadjuvant kjemoterapi er et alternativ til adjuvant kjemoterapi, og anses likeverdig. SSG XX-kriteriene kan ikke benyttes ved neoadjuvant behandling. Hvis neoadjuvant behandling vurderes, anbefales det at malignitetsgrad ≥2, dyp lokalisasjon og tumorstørrelse ≥5 cm brukes som høyrisikokriterier. Inklusjon i en pågående fase II-studie med sekvensiell preoperativ kjemoterapi ved Haukeland universitetssykehus og Oslo universitetssykehus kan vurderes (Sequential Neoadjuvant Chemotherapy in Soft Tissue Sarcoma, 2021-2034).
Anbefaling:
- Pasienter som oppfyller inklusjonskriteriene i SSG XX-studien bør vurderes for adjuvant kjemoterapi med doksorubicin og ifosfamid (Evidensgrad B).
- Neoadjuvant kjemoterapi er et alternativ til adjuvant kjemoterapi for pasienter med høygradig malign tumor, dyp lokalisasjon og tumorstørrelse ≥5 cm.
- Ved lavgradig maligne bløtvevssarkomer anbefales ikke kjemoterapi ved lokalisert sykdom (evidensnivå D).
Abdominalt og retroperitonealt sarkom
Det finnes ingen studier som viser sikker nytte av adjuvant medikamentell behandling ved lokaliserte abdominale eller retroperitoneale bløtvevssarkomer, med unntak av GIST. I tråd med internasjonale retningslinjer er det derfor ikke anbefalt å tilby adjuvant medikamentell behandling. Man kan i spesielle tilfeller, og etter grundig informasjon til pasienten, vurdere å tilby adjuvant kjemoterapi etter tilsvarende prinsipper som for bløtvevssarkomer i ekstremiteter og trunkus, eksempelvis hos unge pasienter med histologiske subtyper som ofte responderer på kjemoterapi (f.eks. synovialt sarkom og UPS) og med høy risiko for tilbakefall etter lokalbehandling. Preoperativ kjemoterapi kan vurderes ved teknisk inoperable svulster som vil kunne opereres ved respons. STRASS 2-studien er en internasjonal, randomisert fase III-studie som har startet rekruttering. Her skal 250 pasienter med høygradig retroperitonealt leiomyosarkom eller dedifferensiert liposarkom randomiseres til henholdsvis neoadjuvant doksorubicin/dakarbazin eller doksorubicin/ifosfamid mot kirurgi alene (Surgery With Our Without Neoadjuvant Chemotherapy in High Risk RetroPeritoneal Sarcoma (STRASS2), 2019-2028). Studien er ikke åpen i Norge.
Gynekologisk sarkom
Det foreligger ingen studier som til nå har vist bedring av overlevelse med adjuvant kjemoterapi ved gynekologiske sarkomer. Man kan i utvalgte tilfeller vurdere adjuvant kjemoterapi eller preoperativ behandling med samme tankegang som i avsnitt over, om Abdominalt og retroperitonealt sarkom, for abdominale og retroperitoneale sarkomer.
Anbefaling:
- Adjuvant kjemoterapi anbefales ikke ved lokalisert abdominalt, retroperitonealt eller gynekologisk bløtvevssarkom (Evidensgrad D).
Spesielle histologiske subtyper
Enkelte histologiske undergrupper av bløtvevssarkom behandles på tilsvarende måte uavhengig av lokalisasjon. For disse undergruppene gjelder ikke anbefalingene gitt i avsnittene over.
Behandling av rhabdomyosarkom følger egne behandlingsprotokoller, og voksne pasienter skal som hovedregel behandles i henhold til pediatriske protokoller. Det brukes spesifikke risikovurderinger og behandlingsopplegg som inneholder kombinasjonskjemoterapi. FaR-RMS protokollen er åpen for rhabdomyosarkompasienter i alle aldre, og anbefales som standardbehandling. Kjemoterapien består her av ifosfamid, vinkristin, aktinomycin D og irinotekan, eventuelt med tillegg av doksorubicin. Etter avsluttet primærbehandling kan det være aktuelt med vedlikeholdsbehandling i form av cyklofosfamid og vinorelbin i lave doser (FaR-RMS: An Overarching Study for Children and Adults With Frontline and Relapsed RhabdoMyoSarcoma (FaR-RMS), 2020-2030). Ved tilbakefall er det aktuelt med behandling i form av vinkristin, irinotekan og temozolomid. Pleomorft rhabdomyosarkom behandles som øvrige bløtvevssarkomer i henhold til avsnitt over, om Bløtvevssarkom i ekstremitet og trunkus, og skal ikke følge FaR-RMS.
Ved andre typer bløtvevssarkomer hos barn følges ofte anbefalinger fra EpSSG basert på NRSTS 2005 studien (A. Ferrari et al., 2021). Det vises her til Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn. (Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn: nasjonal faglig retningslinje, 2020)
Ewing sarkom i bløtvev behandles som Ewing sarkom i ben. For desmoplastisk småcellet rundcellet tumor er det anbefalt å gi tilsvarende behandling som ved Ewing sarkom. Klarcellet sarkom og alveolært bløtdelssarkom er vanligvis ikke følsomme for kjemoterapi, og det anbefales ingen adjuvant behandling. Kjemoterapi ved ekstraskeletalt osteosarkom er omdiskutert. Tradisjonelt har man gitt osteosarkomrettet behandling uten metotreksat, men de siste årene har mange toneangivende sentere internasjonalt gått over til å følge prinsippene for andre bløtvevssarkomer.
Medikamentell behandling av lokalisert bensarkom
Sist faglig oppdatert: 28.02.2022
Osteosarkom
Behandlingen av osteosarkom er multimodal, og som første del av behandlingen gis oftest cytostatika. Ved neoadjuvant kjemoterapi, dvs. preoperativ cytostatikabehandling, oppnås en lokal effekt på tumor som fremmer ekstremitetsbevarende kirurgi, og i tillegg får en mulighet til evaluering av respons på kjemoterapi ved histologisk undersøkelse av operasjonspreparatet.
Fire medikamenter - doksorubicin, metotreksat, ifosfamid og cisplatin - har dokumentert effekt ved osteosarkom, og forskjellige kombinasjoner har vært brukt av ulike grupper internasjonalt (Evidensgrad A) (S. Ferrari et al., 2005). Nytten av modifisering/intensivering av postoperativ kjemoterapi for pasienter med dårlig histologisk respons har blitt testet ut i den internasjonale EURAMOS-1-studien (www.ssg-org.net) (Euramos 1. Randomised trial of the European and American osteosarcoma study group to optimise treatment strategies for resectable osteosarcoma based on histological response to pre-operative chemotherapy, 2012), ved at pasienter med dårlig respons ble randomisert til tillegg av ifosfamid og etoposid til standardregimet med metotreksat, doksorubicin og cisplatin. Tillegg av ifosfamid og etoposid gav ingen overlevelsesgevinst (Marina et al., 2016). Pasienter med god histologisk respons ble randomisert til vedlikeholdsbehandling med interferon-alfa etter avsluttet kjemoterapi eller ingen tilleggsbehandling. Dette baserer seg på studier utført i Stockholm, hvor effekt av interferon-alfa som adjuvans til kirurgi ble antydet (Strander et al., 1995; Strander & Einhorn, 1977). I EURAMOS-1-studien ble det ikke påvist bedre resultater i interferon-armen (S. S. Bielack et al., 2015). Kjemoterapi i henhold til EURAMOS-1-protokollen er anbefalt behandling ved høygradig malignt osteosarkom hos pasienter <40 år (Evidensgrad A). Ved lavgradig osteosarkom anbefales ikke kjemoterapi ved lokalisert sykdom. Basert på resultatene fra EURAMOS-1 benyttes nå samme type kjemoterapiopplegg ved både god og dårlig histologisk respons, nemlig den «korte» behandlingsarmen i studien.
Immunmodulatoren muramyl tripeptid er godkjent av EMA i Europa til bruk adjuvant hos pasienter <30 år med osteosarkom, men anbefales ikke som standard av de ledende osteosarkomgruppene. Muramyl tripeptid er riktignok vist i en randomisert studie i USA å ha effekt på totaloverlevelse (Evidensgrad A), men resultatene er omstridt og diskutert (Marina et al., 2016; Meyers et al., 2008).
Pasienter >40 år var ikke inkluderbare i EURAMOS-1-studien fordi gjennomføring av så intensiv kjemoterapi med høydose metotreksat i denne aldersgruppen som hovedregel er vanskelig. I denne aldersgruppen anbefales kjemoterapi i henhold til EURO-B.O.S.S.-protokollen (S. Ferrari et al., 2018) (Evidensgrad B). Behandlingen her består av doksorubicin, cisplatin og ifosfamid med eller uten metotreksat, og er mindre intensiv enn i EURAMOS-1. Doser og antall kurer må i større grad enn for yngre pasienter tilpasses pasientens alder, komorbiditet og toksisitet underveis i behandlingen.
Kondrosarkom
Ved kondrosarkom er overlevelsen generelt god med kirurgisk behandling alene, og kondrosarkomer er lite følsomme for kjemoterapi. Kjemoterapi blir ikke benyttet rutinemessig, verken adjuvant eller ved metastatisk sykdom. Unntaket er ved dedifferensiert kondrosarkom, der det er aktuelt å tilby kjemoterapi som ved osteosarkom med unntak av metotreksat (Hompland et al., 2021) (Evidensgrad B).
Ewing sarkom
Ewing sarkom er generelt følsom for kjemoterapi, og behandlingen innledes med neoadjuvant kjemoterapi med samme begrunnelse som ved osteosarkom. Behandlingsanbefalingen i Norge er basert på den randomiserte faseIII-studien Euro Ewing 2012 (Evidensgrad A). For alle pasienter består induksjonsbehandlingen av alternerende kurer VDC og IE gitt hver 14.dag, totalt 5 kurer VDC og 4 kurer IE. Foreløpige resultater har vist at induksjonsbehandling med VDC/IE gir bedret overlevelse sammenlignet med VIDE (Strauss et al., 2021). VDC består av vinkristin 2 mg/m2 dag 1, doksorubicin 37,5 mg/m2 dag 1 og 2 og cyklofosfamid 1200 mg/m2 dag 1. IE består av ifosfamid 1,8 g/m2 dag 1-5 og etoposid 100 mg/m2 dag 1-5. Konsoliderende behandling etter lokalbehandling avhenger av risikostratifisering. Pasientene deles inn i tre hovedgrupper:
- Standard risiko. Pasienter med lokalisert sykdom og god histologisk respons på preoperativ kjemoterapi (<10 % viable tumorceller) og initialt tumorvolum <200 ml.
- Høy risiko. Pasienter med lokalisert sykdom og dårlig histologisk respons på preoperativ kjemoterapi (≥10 % viable tumorceller) eller initialt tumorvolum ≥200 ml.
- Metastatisk.
Pasienter med standard risiko får konsoliderende behandling med alternerende IE og VC, totalt 3 kurer IE og 2 kurer VC. Regimet er identisk med det preoperative med unntak av at doksorubicin er tatt bort. Pasienter med høy risiko skal som hovedregel vurderes for høydosebehandling med stamcellestøtte (HMAS), se under. Det gis da én kur VAI postoperativt før man høster stamceller. I enkelte tilfeller kan det bli nødvendig å gi en ekstra kur før BuMel dersom ikke stamcelleproduktet er klart for bruk i tide.
Indikasjon for høydosebehandling med stamcellestøtte
Rasjonalet for HMAS ved Ewing sarkom er å konsolidere en pågående respons på kjemoterapi etter induksjonsbehandling, og at HMAS kan være mer effektivt en konvensjonell kjemoterapi som konsolidering. Det vil altså ikke være aktuelt å vurdere HMAS ved progredierende sykdom eller hvis sykdomsutbredelsen er omfattende på tidspunkt for HMAS.
HMAS ved lokalisert sykdom er undersøkt i to randomiserte studier (Euro-E.W.I.N.G. 99 og Ewing 2008), som er analysert og presentert samlet (Whelan et al., 2018). Pasienter med minst ett av følgende kriterier ble randomisert mellom konsoliderende kjemoterapi med 8 VAI-kurer eller 1 VAI + HMAS med busulfan og melfalan: (i) dårlig histologisk respons på induksjonsbehandling, definert som >10% viable tumorceller i operasjonspreparatet, (ii) dårlig radiologisk respons dersom pasienten ikke gjennomgikk kirurgi, definert som <50% reduksjon av bløtdelskomponenten eller (iii) stort tumorvolum, definert som >200 ml. Kontraindikasjoner til BuMel var strålebehandling som involverte spinalkanalen (dose på minst 30 Gy) eller mot et stort tarmvolum (dose på minst 45 Gy). 78% av pasientene ble inkludert på grunn av dårlig histologisk respons. Pasienter randomisert til HMAS hadde statistisk signfikant bedre event-free survival (EFS) og overall survival (OS). ISG/SSG III-studien var en ikke-randomisert studie der pasienter med lokalisert sykdom og dårlig histologisk eller radiologisk respons (målt ved andre kriterier enn ovenfor) fikk HMAS. Resultatene i denne protokollen tyder også på at HMAS gir bedre overlevelse enn konvensjonell kjemoterapi i den aktuelle pasientgruppen (S. Ferrari et al., 2011). Det finnes ingen randomiserte studier med HMAS etter induksjonsbehandling med VDC/IE. Tross manglende data mener vi at pasienter med dårlig histologisk eller radiologisk respons etter VDC/IE også bør tilbys HMAS. Dokumentasjonen er best når det gjelder pasienter med dårlig histologisk respons som inklusjonskriterium (78% av pasientene), og dette må tas med i vurderingen når man vurderer HMAS hos pasienter med dårlig radiologisk respons eller stort tumorvolum som risikokriterium.
Andre høygradig maligne sarkomer i ben
Udifferensiert pleomorft sarkom oppstått i ben har mange likhetstrekk med osteosarkom og behandles etter de samme retningslinjer. Andre histologiske subtyper kan også oppstå i ben i sjeldne tilfeller, som eksempelvis angiosarkom, leiomyosarkom og synovialt sarkom. Det foreligger ingen tydelige anbefalinger i slike situasjoner, men man vil ofte gi kjemoterapi etter prinsippene ved samme histologiske subtype i bløtvev.
Toksisitet
Behandlingsprotokollene for bensarkom er svært intensive og komplekse, har høy toksisitet og risiko for utvikling av senbivirkninger og krever nøye monitorering. Administrasjon av høydose metotreksat krever spesiell overvåkning og kompetanse. Virkningen av metotreksat motvirkes etter 24 timer ved bruk av folinat kombinert med forsert alkalisk diurese, og utskillelsen av metotreksat via nyrene monitoreres nøye. Ekstra prosedyrer ved forsinket utskillelse og nyresvikt er nøye beskrevet i behandlingsprotokollene.
Anbefaling:
- Kjemoterapi i henhold til EURAMOS-1-protokollens korte arm er anbefalt behandling ved høygradig malignt osteosarkom hos pasienter <40 år (Evidensgrad A).
- Kjemoterapi i henhold til EURO-B.O.S.S.-protokollen er anbefalt behandling for pasienter >40 år med høygradig malignt osteosarkom (Evidensgrad B).
- Ved lavgradig osteosarkom anbefales ikke kjemoterapi ved lokalisert sykdom (evidensnivå D).
- Kjemoterapi anbefales ikke ved lokalisert kondrosarkom, med unntak av ved dedifferensiert kondrosarkom, der det er aktuelt å tilby kjemoterapi i henhold til EURO-B.O.S.S.-protokollen (Evidensgrad B).
- Ewing sarkom behandles i henhold til Euro Ewing 2012 studien (Evidensgrad A).
Gastrointestinal stromal tumor (GIST)
Sist faglig oppdatert: 28.02.2022
Mutasjonsanalyse er obligatorisk når behandling med tyrosinkinasehemmer vurderes, fordi type mutasjon har stor prediktiv verdi for effekt av behandling med tyrosinkinasehemmer.
Adjuvant behandling med imatinib skal vurderes for pasienter med høy risiko for tilbakefall etter modifiserte NIH-kriterier (Joensuu, 2008) (evidensnivå A). Pasienter med veldig lav, lav eller intermediær risiko har svært liten risiko for tilbakefall. I et materiale fra Oslo universitetssykehus er 5-års residivfri overlevelse i disse gruppene >95% (Hølmebakk et al., 2019; Hølmebakk et al., 2018), og det er således ikke grunnlag for adjuvant behandling. Pasienter med høyrisiko GIST bør tilbys 3 års adjuvant behandling med imatinib, basert på en randomisert fase 3 studie utført av SSG og den tyske sarkomgruppen. Pasienter med høyrisiko GIST ble her randomisert mellom 1 år og 3 års behandling med imatinib 400 mg daglig. Studien viste at residivfri overlevelse og totaloverlevelse var bedre ved 3 års adjuvant behandling (Joensuu, Eriksson, et al., 2012; Joensuu et al., 2016; Joensuu et al., 2020). SSG har startet en studie hvor det undersøkes om det er ytterligere gevinst ved å øke behandlingstiden til 5 år (SSG XXII). I vurderingen av indikasjonen for adjuvant behandling må man vektlegge pasientens alder og komorbiditet, sett i lys av at dataene tyder på at man for majoriteten av pasientene utsetter et eventuelt tilbakefall, men ikke forhindrer det. Man bør også ta med mutasjonsanalysen i betraktningen. Pasienter med delesjoner i KIT ekson 11 synes å ha størst nytte av 3 års adjuvant imatinib (Joensuu et al., 2017).
Hvis det har tilkommet tumorruptur, kan man vurdere om pasienten skal behandles som i metastatisk setting, altså med imatinib livslangt. Det foreligger imidlertid ingen konklusive data som støtter dette (evidensnivå D).
Svulster med PDGFRA Asp842Val mutasjon og svulster uten påvist mutasjon i PDGFRA eller KIT har ingen dokumentert effekt av imatinib, og pasienter med disse genotypene skal ikke tilbys imatinib adjuvant (evidensnivå D). Ekson 9 mutasjon er assosiert med dårligere effekt av standard dose imatinib, og man kan vurdere doseøkning til 800 mg x 1 hos pasienter med ekson 9 mutasjon ved metastatisk sydkom. Retrospektive data tyder imidlertid på at det ikke er grunnlag for doseøkning ved adjuvant behandling (Vincenzi et al., 2021).
Ved svulster som er i grenseland for reseksjon, svulster med stor risiko for intraoperativ ruptur og svulster som ved størrelsesreduksjon vil kunne fjernes med et mindre omfattende inngrep, anbefales neoadjuvant behandling med imatinib i 6 til 12 måneder (Casali et al., 2022) (Evidensgrad C). Slik behandling vil være mest aktuell for GIST i øsofagus, cardia ventriculi, duodenum og rektum. Mutasjonsanalyse er normalt en forutsetning for neoadjuvant behandling, men i øvre del av ventrikkelen er over 90 % av GIST følsomme for imatinib, og behandling kan unntaksvis startes uten kjent genotype (Hølmebakk et al., 2021). Ved KIT ekson 9 mutasjon kan doseøkning til 800 mg daglig vurderes. Total behandlingslengde for neoadjuvant og adjuvant behandling skal være totalt 3 år dersom man velger å gi adjuvant behandling i tillegg. Mitosetelling kan ikke brukes til risikoklassifisering etter preoperativ behandling.
Anbefaling:
- Tre års adjuvant behandling med imatinib er anbefalt ved høy risiko for tilbakefall etter modifiserte NIH-kriterier (Evidensgrad A).
- Ved PDGFRA Asp842Val mutasjon og uten påvist mutasjon i PDGFRA eller KIT er adjuvant imatinib ikke anbefalt (Evidensgrad D).
- Neoadjuvant behandling med imatinib i 6-12 måneder kan vurderes ved svulster som er i grenseland for reseksjon, svulster med stor risiko for intraoperativ ruptur og svulster som ved størrelsesreduksjon vil kunne fjernes med et mindre omfattende inngrep (Evidensgrad C).
Støttebehandling og rettigheter
Ernæring og sarkombehandling
Sist faglig oppdatert: 28.02.2022
Kreftsykdom og kreftbehandling gir ofte redusert matlyst og vekttap. En kreftpasient som spiser mindre enn behovet sitt, og som taper vekt, vil tape en større andel muskelmasse enn fettvev i forhold til en frisk person som slanker seg. Dette er uheldig fordi pasienten kan få mindre kapasitet til å metabolisere kjemoterapi og økt toksisitet av behandlingen. Underernæring og muskeltap vil også føre til:
- Redusert immunforsvar
- Redusert effekt av behandlingen
- Redusert sårtilheling etter kirurgi
- Redusert subjektiv opplevelse av livskvalitet
- Lavere aktivitetsnivå
- Redusert overlevelse
De fleste kreftpasienter vil ha behov for ernæringsintervensjon i løpet av behandlingsforløpet og ved progresjon av sykdommen. Det er anslått at 40 % av alle kreftpasienter er i ernæringsmessig risiko (Tangvik et al., 2015). For å oppnå best mulig resultat av kreftbehandlingen og best mulig livskvalitet hos pasienten, er det viktig å gi ernæringsbehandling parallelt med kreftbehandlingen. Dette er en del av pasientens rehabilitering og gjennomføres ved et samarbeid mellom lege, klinisk ernæringsfysiolog, sykepleier, hjelpepleier og postvert.
Det finnes ingen universell definisjon på eller diagnostiske kriterier for underernæring. I litteraturen defineres ofte underernæring som en ernæringssituasjon hvor utilstrekkelig inntak eller opptak av energi, protein og andre næringsstoffer gir målbare negative effekter på kroppssammensetning, -funksjon og klinisk utfall. Helsedirektoratets «Nasjonale faglige retningslinjer for forebygging og behandling av underernæring» har foreslått at pasienter er underernært hvis de oppfyller minst ett av kriteriene under (Nasjonale faglige retningslinjer for forebygging og behandling av underernæring, 2009):
- Ufrivillig vekttap > 10 % siste 3–6 md. eller > 5 % siste 2 md.
- KMI < 18,5 kg/m2 (> 70 år: KMI < 20 kg/m2)
- KMI < 20 kg/m2 (> 70 år: KMI < 22 kg/m2) og samtidig ufrivillig vekttap > 5 % siste 6 md.
- Matinntak < 50 % av beregnet behov siste uke
Regelmessig vurdering av ernæringsstatus er en viktig del av kreftbehandlingen. Hensikten er å identifisere ernæringsmessig risiko og underernæring tidlig, for raskest mulig å iverksette tiltak. Ernæringsstatus hos voksne pasienter kan klassifiseres ved hjelp av Nutrition Risk Score (NRS 2002) eller for en nøyere kartlegging Subjective Global Assessment (SGA). Vurderingen bør også inkludere ernæringsmessig risiko i forhold til behandlingen som skal gis, for eksempel om det er forventet kvalme eller munnsårhet i forbindelse med behandlingen.
Før tiltak settes i gang bør underliggende faktorer til redusert matlyst (kvalme, obstipasjon, dysfagi, smerte, psykososiale faktorer o.s.v) utredes og behandles. Pasienten bør så tilbys næringstett mat, drikke og mellommåltider. Hvis en ikke kommer i mål med dette må man supplere matinntaket med næringsdrikker og eventuelt enteral- og/eller parenteral ernæring. Næringsdrikker og enteral ernæring er å foretrekke dersom mage-tarmkanalen fungerer.
For kreftpasienter i vekst (under 18 år) kan underernæring forekomme på tross av stabil vekt eller ved små vekttap. Pasientens høyde bør måles hver 3. måned. Vekt og høydeutvikling føres inn i vekstkurver. Underernæring (og overernæring) identifiseres ved forandringer i vekstkurver tilsvarende 1–2 percentiler.
Tommelfingerregel for beregning av energibehov hos kreftpasienter (Arends et al., 2017; Nasjonale faglige retningslinjer for forebygging og behandling av underernæring, 2009):
- Vedlikehold av vekt for oppegående pasienter, 30–35 kcal/kg kroppsvekt/dag
- Vedlikehold av vekt for sengeliggende pasienter, 20–25 kcal/kg kroppsvekt/dag
- Vektøkning, 40 kcal/kg kroppsvekt/dag
- Proteinbehov, 1,0–1,5 g/kg kroppsvekt/dag
I forkant av store operasjoner har alvorlig underernærte pasienter fordel av 10–14 dager med ernæringsstøtte, selv om operasjonen må utsettes. Ernæringsstøtte bør gis per oralt eller enteralt. Hvis ikke dette blir tilstrekkelig, må man benytte parenteral ernæringsstøtte (7–10 dager). Ernæring inngår som en del av den perioperative behandlingsplanen, for eksempel når Enhanced Recovery After Surgery, ERAS, følges (Gustafsson et al., 2013).
Pasienter som har stabil vekt under sykdom og behandling bør ha et variert matinntak med mye frukt, bær, grønnsaker, fisk og grove kornprodukter, i tråd med de offentlige anbefalingene for den generelle befolkningen.
Sykepleie til pasienter med sarkom
Sist faglig oppdatert: 28.02.2022
Utredningsfasen
De første symptomene hos pasienter med sarkom er ofte smerte og hevelse. Varme og ømhet kan også forekomme, men ellers er pasientens allmenntilstand ofte upåvirket. Er pasienten ung kan symptomene ofte forveksles med andre godartede tilstander, og familien kan ofte ha brukt lang tid på å få riktig diagnose og komme til et spesialistsykehus. Den lokale helsetjenesten har sjelden kontakt med denne type svulster og pasienter, og pårørende kan sitte med en følelse av at de ikke blir tatt på alvor. Dette kan føre til skyldfølelse hos både pasient og pårørende som føler at de burde ha stått på mer for å få en riktig diagnose på et tidligere tidspunkt. Ved hjelp av god informasjon kan helsepersonell hjelpe pasienten og/eller pårørende til å unngå skyldfølelse, og dermed lettere støtte dem i å fokusere på utredning og behandling.
Når pasienten kommer til spesialistsykehus er det mange forberedende undersøkelser. Mange barn og ungdommer opplever smerten som oppstår i forbindelse med prosedyrer og prøvetakinger som den verste smerten. Ved å være godt informert på forhånd kan pasienten oppleve å ha kontroll over situasjonen under undersøkelsene. Sykepleieren har en viktig funksjon i å informere om hva som skal skje, og planlegging i forhold til bruk av pasientens tid. Pasienten må på forhånd få muntlig og skriftlig informasjon om alle undersøkelser slik at pasienten opplever å ha kontroll over egen situasjon. Informasjonen må gis i rolige omgivelser hvor pasienten føler at han/hun har mulighet til å stille spørsmål.
Helsepersonell må forsikre seg om at pasienten er smertelindret før de forskjellige undersøkelsene skal gjennomføres. Å være godt informert på forhånd kan gjøre at pasienten bedre takler smerte.
Hele familien er i denne perioden utsatt for en stor mental belastning og det stilles store krav til den informasjonen familien har behov for i utredningsfasen.
Behandlingsfasen
Når det er klart at diagnosen er sarkom, starter behandlingen så raskt som mulig. Behandlingen kan bestå av bare operasjon eller operasjon i kombinasjon med kjemoterapi og/eller strålebehandling.
Cellegiftbehandlingen vil for sarkompasienten være langvarig. Hva hver enkelt pasient ønsker å vite vil variere, og dette bør kartlegges. Det må informeres om de akutte bivirkningene, som for eksempel håravfall, kvalme, såre slimhinner og infeksjoner, og også hvilke tiltak pasienten selv kan gjøre for å lindre plagene. Mange av cellegiftene sarkompasienten får gir benmargstoksisitet, og dette medfører ofte infeksjoner mellom kurene. Pasienten bør informeres både skriftlig og muntlig om hvordan han skal forholde seg i den neutropene fasen, og hva han skal gjøre hvis feber oppstår.
Mange pasienter, og spesielt ungdommer, er opptatt hvordan de vil se ut under og etter behandlingstiden. Mange vil oppleve å gå ned i vekt, få arr på kroppen, og pasienter med sarkom skal som oftest gjennomgå kirurgi for å få fjernet svulsten. Hvor omfattende operasjonen blir er avhengig av tumors størrelse og lokalisasjon. Dette vil ofte medføre en varig funksjonsendring/handikap. I denne fasen har sykepleier, sammen med fysioterapeut, ansvar for at pasienten så tidlig som mulig etter operasjonen begynner med trening for å gjenvinne styrke og funksjon, eller finne nye måter å klare seg selv på. Dette vil være med på å gjøre pasienten kjent med, og trygg på, sitt nye kroppsbilde.
En ekstra utfordring for pasienter med sarkom er at mange skal ha mange cellegiftkurer også etter operasjonen, og det vil gjøre gjenopptreningen vanskeligere på grunn av de bivirkninger cellegiften gir.
At pasienten uttrykker frustrasjon over ikke å mestre ting som tidligere, er ikke uvanlig. Ved å våge å være til stede også når pasienten viser følelser, og vise aksept overfor individuelle måter å utrykke seg på, vil helsepersonellet kunne bidra til aksept av mennesket som er rammet, og aktivt være til støtte og hjelp på veien videre. Siden sarkom kan gi varige fysiske handikap, er det avgjørende at sykepleier tar del i pasientens hverdag på sykehuset. Gjennom tillit, kontinuitet og samtale bidrar sykepleier til å øke pasientens opplevelse av mestring av egen livssituasjon. Dette er igjen avgjørende for rehabiliteringen og mulighetene for at pasienten skal kjenne seg fornøyd med kroppen etter operasjonen.
For pasienter som gjennomgår spesielle operasjoner som amputasjoner eller rotasjonsplastikk, kan det være betydningsfullt å få møte pasienter som tidligere har gjennomgått samme operasjon, for lettere å se mulighetene med de begrensningene man har fått på grunn av operasjonen. Her kan sykepleierne være et viktig bindeledd.
Ungdommer har ofte fokus på aktiviteter, venner, utseende, seksualitet, skolegang og fremtiden. Det samme fokus gjelder selv om ungdommen har fått et sarkom, men ungdommen må nå forholde seg til at kroppen forandres som følge av behandlingen. For at ungdommen skal kunne fortsette å ha det samme fokus, er det viktig at det legges til rette for at ungdommen kan fortsette å ha kontakt med venner og skole. Dette kan gjøres ved å legge til rette for besøk og overnatting for venner på avdelingen. Ofte kan det å ha en venn på besøk i stedet for mor eller far være en god opplevelse for ungdommen. Ved å få bekreftelse fra venner på at ungdommen fortsatt er en del av gjengen, til tross for forandringer i utseendet, kan dette gjøre det lettere for ungdommen å godta sitt nye kroppsbilde. Et skolebesøk hvor en sykepleier er til stede, kan være med på å avmystifisere mye rundt diagnose og behandling, og gjøre det mindre skummelt for ungdommer å komme på besøk til sykehuset.
Oppfølgingsfasen
Etter å ha gjennomgått et langt behandlingsopplegg for sitt sarkom, skal den enkelte pasient inn i et langt – og for noen – livslangt kontrollopplegg. Kontrollene vil i stor grad foregå ved poliklinikker på spesialsykehus her i Norge. For pasienten innebærer det lengre reiser, samtidig som de opplever stor trygghet ved at de møter de samme menneskene ved kontrollene hver gang. Sykepleierens oppgave vil her være å bidra til fortsatt kontinuitet og bindeledd i kontrollopplegget. Mange sykehus praktiserer pasientansvarlig sykepleie slik at pasienten til enhver tid vet hvem de skal henvende seg til dersom endringer/uklarheter oppstår. En del pasienter opplever det mindre skummelt å dele problemer med sykepleieren enn med legen. Å komme til kontroll betyr at det blir en kort visitt på sykehuset, og det er viktig at sykepleieren kommuniserer til pasienten at hun har tid til samtale hvis pasienten har behov for det. Dette må gjøres både verbalt og nonverbalt. Noen pasienter kan ha behov for ekstra støtte etter at behandlingen er avsluttet. Behovet kan være å få snakke igjennom sykdomstiden og hva den har medført, for å kunne gå videre i livet. Hvis pasienten ikke gis mulighet til å bearbeide dette kan det gi pasienten problemer senere i livet.
Rehabilitering av sarkompasienten starter ved diagnosetidspunktet, og er i fokus gjennom hele behandlingsforløpet og videre gjennom kontrollopplegget. Ofte jobber alle profesjoner i team med sarkompasienten, og sykepleier er her en viktig koordinator.
Allmennlege/pasientens fastlege har en viktig rolle i dette teamet når det gjelder oppfølging etter behandling, både på kort og lengre sikt. Ved avslutning av behandlingen skal sykehuset informere allmennlege/fastlege om mulige senskader man bør være årvåken for.
Fysioterapi til pasienter med sarkom
Sist faglig oppdatert: 28.02.2022
Et viktig mål for fysioterapi innen onkologi er å optimalisere funksjon samt forebygge og lindre ulike følgetilstander av sykdom og behandling. Sarkompasienter har høy risiko for å utvikle fysisk funksjonssvikt av en eller annen art, og bør henvises til fysioterapeut.
Diagnose og oppstart av behandling
Sarkombehandling er langvarig og sammensatt og kan føre til store endringer i pasientens liv. En omfattende behandling fører ofte til generelle og spesifikke fysiske funksjonsvansker.
Ved oppstart av behandlingen er det viktig med en kartlegging av risikopasienters (defineres nærmere av lege) fysiske funksjonsnivå. Generelle og spesifikke muskelstyrketester, bevegelighetstester, balansetester, sensibilitetsundersøkelser, lungefunksjonsprøver (spirometri), smerteproblemer og testing av utholdenhet, bør inngå i kartleggingen (Evidensgrad D).
Rehabiliteringsrettede tiltak bør iverksettes så snart behovet er avdekket. Det tverrfaglige teamet bør vurdere å utarbeide en individuell rehabiliteringsplan (IP). Fysioterapeuten gir råd og veiledning i forhold til fysisk aktivitet/trening under selve behandlingsperioden, og utarbeider et tilpasset treningsprogram. Sammen med andre medlemmer i det tverrfaglige teamet kan fysioterapeuten bidra til at pasienten opplever å mestre livssituasjonen (Evidensgrad D).
Kirurgiske inngrep med risiko for funksjonssvikt
Preoperativ fase
Fysioterapeuten samarbeider med lege og sykepleier om informasjon vedrørende operasjon og etterbehandling. Pasienter som får informasjon om hva postoperativ fysioterapi vil innbefatte, og trener om mulig på forflytningsmetoder og de spesifikke øvelsene som kan bli aktuelle etter operasjonen, får mindre angst før operasjon og kommer seg raskere etter operasjon (Giraudet-Le Quintrec et al., 2003) (Evidensgrad A).
Postoperativ fase
Behov for fysioterapi vil variere med omfang av operasjonen. Noen sarkompasienter skal ha daglig fysioterapi, og ofte flere ganger pr dag. Det skal foreligge skriftlig henvisning fra ortoped som gir klare retningslinjer og kontraindikasjoner i forhold til mobilisering, belastning og øvelser. Fysioterapien krever spesiell tilnærming og individuelle tiltak som stadig må justeres. Et nært samarbeid med pasient, lege, sykepleier og annet personale er nødvendig.
Pasienter som gjennomgår omfattende operasjoner står i fare for å utvikle respiratoriske komplikasjoner som atelektase og pneumoni (Physiotherapy for respiratory and cardiac problems: adults and paediatrics, 2008). Etter større kirurgi er det ofte behov for lungefysioterapi inntil lungestatus er tilfredsstillende (Hough, 2001) (Evidensgrad A).
Det er høy risiko for trombose hos kreftpasienter generelt, og spesielt for dem som gjennomgår stor kirurgi (A. Y. Lee et al., 2005). Dyp venetrombose kan forebygges medikamentelt og/eller ved bruk av antitrombosestrømpe (Elvsaas, Graff, Harboe, & Norderhaug, 2008). I samarbeid med lege og sykepleier tilpasser fysioterapeuten antitrombosestrømper, instruerer i forebyggende øvelser og mobiliserer pasienten etter gitte retningslinjer (Evidensgrad A).
Ved subkutane sarkom fjernes tumor, hud, subcutis og fasie. Dette gir vanligvis liten funksjonell påvirkning. Aktuell fysioterapi kan være å kontrollere bevegeligheten, spesielt ved leddnære operasjoner, samt tøyningsøvelser og generell kondisjonstrening.
Ved dypt beliggende sarkom blir inngrepet mer omfattende. Tumor, og en eller flere muskler, fjernes, og rekonstruktiv kirurgi kan være nødvendig. En studie viser at fjerning av en enkel hoftemuskel eller lårmuskel ikke fører til noe nevneverdig tap av funksjon (Markhede & Stener, 1981). Studien viser også at funksjonell svekkelse etter å ha fjernet flere muskler for en stor grad vil bli kompensert for av hypertrofi av gjenværende muskler med samme funksjon. Aktuell fysioterapi kan være generell og spesifikk funksjonstrening, samt tilpassing av hjelpemidler og ortoser.
Ved Ewing sarkom og osteosarkom kan operasjonen være svært omfattende, med for eksempel innsetting av spesialprotese og rekonstruktiv kirurgi. Det er svært viktig med klare retningslinjer og kontraindikasjoner fra ortopeden i forhold til mobilisering, belastning og øvelser. Noen ganger er amputasjon nødvendig. Aktuelle fysioterapitiltak er generell opptrening, trening med protese, samt tilrettelegging av hjelpemidler.
Sarkompasienter skal ofte ha postoperative cellegiftkurer og dette vil forsinke og komplisere gjenopptreningen.
En studie av helse blant langtidsoverlevere med bensarkom tyder på at pasientene får akseptabel til god fysisk funksjon og god livskvalitet (Aksnes, 2008).
Spesielle utfordringer ved multimodal behandling av sarkom
Fatigue
Fatigue er et hyppig forekommende symptom hos sarkompasienter. Følelsen av utmattelse og kraftløshet er plagsom og invalidiserenede. Tilpasset fysisk trening er gunstig og gir bedret livskvalitet (Cramp & Daniel, 2008) (Evidensgrad A).
Smerter
Både cellegiften, den ortopediske operasjonen og ev. strålebehandling, kan forårsake perifer polynevropati og andre nevrogene smertetilstander. Fysioterapitiltak som innebærer sansemotorisk stimulering og øvelser kan lindre plagene. Ulike former for avspenningsteknikker kan være positivt.
Sentrale nevrologiske utfall
Sentrale nevrologiske utfall kan oppstå som en komplikasjon til cellegiften. Fysioterapeuten må straks melde fra til vakthavende onkolog hvis pasienten har slike symptomer.
Svekket hjertemuskulatur
Cellegiften doksorubicin hører til gruppen antracykliner og virker toksisk på hjertemuskulaturen. Det kan være restriksjoner på intensiteten i utholdenhetstrening. Dette må avklares i hvert enkelt tilfelle i samarbeid med onkolog.
Lave blodverdier
Leukopeni og trombocytopeni opptrer regelmessig mellom kurene og kan blant annet føre til at pasienten må isoleres. Fysisk trening kan være kontraindisert ved høy grad av generell sykdomsfølese, høy feber og/eller svært lave trombocytter. Grad av fysisk aktivitetsnivå avtales i samarbeid med onkolog.
Osteoporose og fare for mikrofrakturer
Både cellegiften, andre medisiner, inaktivitet/langvarig avlastning og ikke minst strålebehandling, disponerer for osteoporose. Det er viktig at fysioterapeuten er klar over dette i forbindelse med treningen og når pasienten skal øke belastningen.
Ødemer
Ulike former for ødem kan ramme sarkompasienter, og aktuelle fysioterapitiltak som f.eks. komplett manuell lymfedrenasje settes i verk.
Se også www.lymfoedem.no.
Se også Nasjonalt Handlingsprogram for lymfomer (Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne lymfomer, 2012).
Overføring til primærhelsetjenesten eller rehabiliteringsinstitusjon
Ved overføring til primærhelsetjenesten eller rehabiliteringsinstitusjon, skriver fysioterapeuten epikrise og legger ved kopi av operasjonsbeskrivelsen. Eventuelle retningslinjer/kvalitetsdokument legges også ved. Telefonisk kontakt kan i tillegg være nødvendig.
Poliklinisk kontroll
Ved utskriving får pasienten time til kontroll hos ortoped og onkolog. Fysioterapeut kan delta ved kontrollene og gi pasienten en nærmere oppfølging samme dag.
Fysioterapi ved palliativ behandling
Kreftpasienter med langtkommen sykdom vil gradvis få redusert fysisk funksjon (Cramp & Daniel, 2008). Trening for palliative pasienter viser positiv effekt både på objektive funksjonsmål og symptomer som fatigue, kvalme og angst samt livskvalitet (Jordhøy, Fayers, Loge, Ahlner-Elmqvist, & Kaasa, 2001).
Se også Nasjonalt Handlingsprogram for palliasjon i kreftomsorgen (Nasjonalt handlingsprogram for palliasjon i kreftomsorgen: nasjonal faglig retningslinje, 2019).
Psykososiale forhold: kartlegging/støtte/behandling
Sist faglig oppdatert: 28.02.2022
Sosionomtjenestens målsetting er å motvirke, eller minske psykososiale konsekvenser av kreftsykdom og behandling for pasienter og pårørende.
Sarkom er en kreftsykdom som rammer personer i alle aldre. Flere er skoleelever/studenter i starten av sitt yrkesliv og voksenliv. Mange er i yrkesaktiv alder hvor noen også er foreldre til barn under 18 år.
Sykdom og behandling kan få konsekvenser for både pasient og pårørende. Den kan føre til følelsesmessige reaksjoner som sorg, krise, angst og depresjon. Den kan innvirke på relasjonelle forhold til ektefelle, barn og sosialt nettverk. Den kan få konsekvenser for utdannelse/arbeidsliv, økonomi og levekår, samt innvirke på åndelige og eksistensielle forhold.
De senere årene har også de pårørendes situasjon fått økende oppmerksomhet både i forskning og i klinikken. Barn som pårørende har fått lovfestede rettigheter. (Se kapittel Pårørendeveileder.)
Hvilke psykososiale bekymringer/problemer som kan oppstå vil være avhengig av:
- Sykdommens alvorlighetsgrad
- Behandlingens varighet
- Om behandlingen har kurativt eller palliativt siktemål
- Pasienten/familiens nettverk og ressurser
Det bør allerede på diagnosetidspunktet gjøres en psykososial kartlegging av pasienten og familiens situasjon. Målsettingen er å:
- Forebygge at problemer oppstår ved å gi informasjon, råd og veiledning om mestringsstrategier, om aktuelle rettigheter og offentlige hjelpe- og støtteordninger
- Fange opp risikopasienter/familier
- Avdekke behov for hjelp både i forhold til det følelsesmessige, praktiske, økonomiske, fysiske og sosiale.
- Tilby tilrettelegging for individuell plan
- Formidle kontakt med hjelpeinstanser i pasientens hjemkommune
Sosionomer i sykehus kan være aktuelle fagpersoner til å foreta en slik kartlegging. De har spisskompetanse og bred kunnskap om rettigheter, hjelpe- og støtteordninger, og kan gi adekvat informasjon, råd og veiledning. Sosionomer har også kompetanse i nettverks- og familiearbeid, de har kunnskap om lovverk og erfaring i å bruke ulike hjelpeinstanser i og utenfor sykehus. Det kommunale hjelpeapparatet er til dels svært oppdelt med forskjellig organisering. Det er derfor nødvendig å undersøke, og skreddersy, hjelpetilbudet for den enkelte pasient og familie.
Det er viktig å ha både et kortsiktig og et langsiktig perspektiv for det psykososiale arbeidet. Allerede på diagnosetidspunktet bør en ha tanker/planer for pasientens rehabilitering og tiltak som skal til for at pasienten og familien skal komme best mulig tilbake til dagliglivet.
Det psykososiale arbeidet forutsetter tverrfaglig samarbeid. Aktuelle yrkesgrupper i tillegg til behandlingspersonalet som lege og sykepleier, kan være sosionom, fysioterapeut, ergoterapeut, ernæringsfysiolog, psykiater, psykolog, psykiatrisk sykepleier og prest.
Det er en forutseting at det psykososiale arbeidet er et tilbud, og at det utføres ut fra pasienten og familiens ønsker og behov (brukerperspektivet).
Viktige rettigheter/hjelpeordninger
Grunn- og hjelpestønad
Det er en utbredt oppfatning at grunn- og hjelpestønad er en rettighet ved kreftsykdom. Disse rettighetene er imidlertid svært begrenset til spesielle utgifter og situasjoner. De har et varighetskrav til sykdommen på 2–3 år. Stønadene er skattefrie og utbetales månedlig.
Grunnstønad
Grunnsøknad skal helt eller delvis dekke visse typer ekstrautgifter som har oppstått pga. sykdom, og som friske personer ikke har. Ekstrautgiftene må være løpende, dvs. stadig tilbakevendende. De mest aktuelle ekstrautgifter for kreftpasienter er utgifter til transport, støttebandasjer/strømper, ekstra slitasje på klær/sengetøy. Ekstrautgiftene og behovet må dokumenteres.
Hjelpestønad
Hjelpestønad kan ytes dersom pasienten på grunn av sykdom har et særskilt behov for tilsyn, pleie og omsorgsbehov. Det er et vilkår at det foreligger et privat pleieforhold. Ved vurdering av hjelpebehovet kan det også legges vekt på stimulering, opplæring og trening som utføres i hjemmet.
Forhøyet hjelpestønad
Funksjonshemmede eller alvorlig syke barn under 18 år som har et betydelig behov for ekstra tilsyn og pleie kan få forhøyet hjelpestønad. Ved vurdering av forhøyet hjelpestønad, og hvilken sats som skal anvendes, legges det vekt på hvor mye barnet/ungdommens fysiske og psykiske funksjonsevne er nedsatt, omfanget av tilsyn og pleie, behovet for stimulering, opplæring og trening samt hvor mye pleieoppgaven binder den som gjør arbeidet.
Barn/ungdom med kreftsykdom kan ha krav på grunnstønad og forhøyet hjelpestønad etter ovennevnte retningslinjer selv om behandlings- og rehabiliteringstiden er av kortere varighet.
Søknadsskjema og mer utførlig omtale av grunn- og hjelpestønad finnes i folketrygdloven kap. Kirurgisk behandling av lokalisert sykdom, www.nav.no.
Pleiepenger ved alvorlig sykdom hos barn/ungdom under 18 år
Den som har omsorg for et alvorlig sykt barn eller barn som er innlagt i helseinstitusjon, har rett til pleiepenger. Pleiepenger utbetales etter de samme reglene som sykepenger ved egen sykdom. Det er en forutsetning at foreldrene av hensyn til barnet må oppholde seg i helseinstitusjonen eller være hjemme, fordi barnet trenger kontinuerlig tilsyn og pleie. Lov om folketrygd § 9‑11 (Lov om folketrygd (folketrygdloven)). Søknadsskjema og mer utførlig informasjon www.nav.no.
Arbeidsavklaringspenger
Arbeidsavklaringspenger kan være en aktuell ytelse når retten til sykepenger opphører etter 52 uker, eller når pasienten ikke har rett til sykepenger. Stønaden skal sikre inntekt i en overgangsperiode hvor pasienten har behov for medisinsk behandling, eller annen oppfølging fra NAV, for å komme i arbeid.
Pasienter som ikke har hatt lønnsinntekt eller andre ytelser før arbeidsevnen ble nedsatt, vil kunne motta arbeidsavklaringspenger tilsvarende en årlig minsteytelse fra første sykedag.
Pasienter som fyller vilkårene for både sykepenger og arbeidsavklaringspenger, har rett til å velge ytelsen som gir høyest utbetaling.
Studenter som har behov for aktiv behandling for å gjenoppta studier og som ikke har rett til stipend under sykdom fra Lånekassen, vil kunne søke arbeidsavklaringspenger fra første sykedag. Søknadsskjema og mer uførlig informasjon www.nav.no.
Statens lånekasse kan gjøre om lån til stipend i inntil 4½ måneder ved sykdom. Informasjon og søknadsskjema finnes på: www.lanekassen.no.
Oppfølging og etterkontroll etter avsluttet kurativ behandling
Oppfølging og etterkontroll
Sist faglig oppdatert: 28.02.2022
Etterkontroll av sarkom
Oppfølging av pasienter som har vært behandlet med kurativt siktemål for et sarkom har som formål:
-
- Å oppdage tilbakefall av sykdommen (metastaser eller lokalt residiv).
- Å kartlegge eventuelle seneffekter etter behandlingen og iverksette nødvendige tiltak.
Anbefalt hyppighet og varighet av kontrollene er veiledende. Pasienter kan følges opp med både tettere og sjeldnere kontroller basert på individuell vurdering.
Kontrollene etter behandling for sarkom utføres primært ved et sarkomsenter på grunn av krav til spesiell radiologisk, ortopedisk og onkologisk kompetanse, samt fortløpende nøyaktig oppdatering av kvalitetsregisteret. I enkelte tilfeller (for eksempel på grunn av alder, geografisk avstand eller pasientens mobilitet), vil kontrollene helt eller delvis kunne overføres til pasientens lokalsykehus eller fastlege. Dette forutsetter at oppfølgingsnotater sendes det aktuelle sarkomsenter. Det ansees også formålstjenlig å samarbeide med lokale onkologiske og kirurgiske avdelinger slik at kompetansen heves og samarbeidet vektlegges.
Pasienter som har fått kompliserte ortopediske rekonstruksjoner (for eksempel endoproteser), trenger langvarig – i noen tilfeller livslang – oppfølging.
Bløtvevssarkom i ekstremiteter og trunkus
Lavgradig maligne bløtvevssarkom
Antall kontroller per år:
- Hver 6. måned 1–5 år
- Årlig i 6–10 år (individuell vurdering)
Undersøkelser:
- Klinisk undersøkelse, spesielt av primærtumorområdet
- Røntgen thorax, eventuelt MR av primærtumorområdet
- CT thorax ved usikre eller metastasesuspekte funn ved røntgen thorax
Høygradig maligne bløtvevssarkom
Antall kontroller per år:
- Hver 3. måned 1 år
- Hver 4. måned i 2–3 år
- Hver 6. måned 4–5 år
- Årlig i 6–10 år
Undersøkelser:
- Klinisk undersøkelse, spesielt av primærtumorområdet
- Røntgen thorax, eventuelt MR av primærtumorområdet
- CT thorax ved usikre eller metastasesuspekte funn på røntgen thorax
- CT thorax ved hver annen kontroll av pasienter i remisjon etter lungemetastatisk sykdom
Pasienter som har fått kjemoterapi
- Blodprøver inkludert elektrolytter, lever og nyrefunksjon ved hver kontroll.
- GFR (nyrefunksjonsundersøkelse) og ekko cor (evt MUGA), 1, 5 og 10 år etter behandling.
Bensarkom
Lavgradig maligne bensarkom
Antall kontroller per år:
- Hver 6. måned 1–5 år
- Årlig i 6–10 år (valgfritt)
Undersøkelser:
- Klinisk undersøkelse, spesielt av primærtumorområdet
- Røntgen thorax, røntgen av primærtumorområdet med ev. MR
- CT thorax ved usikre eller metastasesuspekte funn ved røntgen thorax
Høygradig maligne bensarkom
Antall kontroller per år:
- Hver 3. måned i 1 år
- Hver 4. måned 2–3 år
- Hver 6. måned 4–5 år
- Årlig i 6–10 år
Undersøkelser:
- Blodprøver inkludert elektrolytter, lever og nyrefunksjon ved hver kontroll
- Klinisk undersøkelse, spesielt av primærtumorområdet
- Røntgen thorax, eventuelt røntgen av primærtumorområdet
- CT thorax ved usikre eller metastasesuspekte funn på røntgen thorax
- CT, MR og/eller FDG PET/CT ved mistanke om metastaser til andre organer (liberal indikasjon)
- CT thorax ved hver annen kontroll av pasienter i remisjon etter lungemetastatisk sykdom
Pasienter som har fått kjemoterapi
- Blodprøver inkludert elektrolytter, lever og nyrefunksjon
- GFR (nyrefunksjonsundersøkelse) og hjertefunksjonsundersøkelse (ekko cor, evt MUGA) etter 1, 5 og 10 år.
Retroperitonealt, abdominalt og gynekologisk sarkom
Det foreligger ingen data som viser nytteverdien av systematiske kontroller, og heller ikke hva som er optimalt oppfølgingsintervall eller optimal varighet av kontroller.
Opererte pasienter kontrolleres med klinisk undersøkelse (inkludert GU og vaginal ultralydundersøkelse ved gynekologisk sarkom), røntgen. evt. CT thorax og CT abdomen/bekken.
Høygradig sarkom: Hver 6. måned i 5 år, deretter årlig i ytterligere 5 år.
Lavgradig sarkom: Første kontroll etter 6 måneder, deretter årlig i 5–10 år. Intervallene kan vurderes økt til 2 år (Gronchi et al., 2021).
GIST: Det er svært liten risiko for tilbakefall ved GIST klassifisert som meget lav, lav eller intermediær risiko etter modifiserte NIH-kriterier. Data fra Oslo Universitetssykehus, Radiumhospitalet, viser at 5-års residivfri overlevelse er >90% i alle disse gruppene (Hølmebakk et al., 2018; Upubliserte data), hvilket samsvarer godt med internasjonale publikasjoner (Bischof et al., 2015; Joensuu, Vehtari, et al., 2012). Anbefalinger om rutinemessige kontroller følger under, men det presiseres at individuelle vurderinger bør gjøres. Det kan være aktuelt både å avstå fra rutinemessige kontroller ved eksempelvis høy alder eller alvorlig komorbiditet, og å øke hyppighet eller varighet av kontroller basert på individuelle risikovurderinger. Det bør være lav terskel for bildediagnostikk ved symptomer som kan være assosiert med tilbakefall.
- GIST med meget lav, lav eller intermediær risiko etter modifiserte NIH-kriterier: Ingen kontroll.
- GIST med høy risiko etter modifiserte NIH-kriterier:
- Hver 6. måned under adjuvant behandling og i 5 år etter avsluttet adjuvant behandling. Hyppigere kliniske kontroller kan være nødvendig under adjuvant behandling grunnet bivirkninger av imatinib.
- Hver 6. måned i 5 år etter operasjon for pasienter uten adjuvant behandling.
Sarkom hos barn
Barn som er inkludert i kliniske studier følges i henhold til den aktuelle protokoll. For øvrige pasienter følges hovedsakelig anbefalingene i avsnittene over, men kan tilpasses etter individuelle vurderinger. Barn som kureres for sarkom har mange år igjen å leve. Det er derfor svært viktig at de følges opp med tanke på seneffekter av behandlingen, slik at seneffektene kan forebygges og eventuelt behandles. Det vises til Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn (Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn: nasjonal faglig retningslinje, 2020) for ytterligere detaljer angående oppfølging av barn og ungdom.
Seneffekter
Sist faglig oppdatert: 28.02.2022
Mange sarkompasienter har mottatt omfattende behandling, i form av store kirurgiske inngrep, strålebehandling og intensiv kjemoterapi. Risikoen for ulike typer av seneffekter er således betydelig. Helsedirektoratet har utgitt rapporten «Seneffekter etter kreftbehandling (Seneffekter etter kreftbehandling, 2020), og det henvises til denne for utfyllende litteratur om temaet. Her omtales ulike typer seneffekter, inkludert spesielle forhold etter kreftbehandling hos barn.
Behandling av metastatisk sykdom / livsforlengende og palliativ behandling
Kirurgi
Sist faglig oppdatert: 28.02.2022
Behandling av lokalt residiv ved bløtvevssarkom
Grundig utredning er viktig, og den kirurgiske behandlingen vil styres av residivets anatomiske utbredelse, malignitetsgraden, og eventuelle metastaser. Hvis man har virksom kjemoterapi eller dersom pasienten ikke har fått strålebehandling, kan disse modaliteter være aktuelle. Lokal eksisjon av residivet, eller amputasjon, kan være aktuell behandling.
Behandling av lokalt residiv ved bensarkom
Behandlingen må styres av residivets anatomiske utbredelse, tumors malignitetsgrad og eventuelle metastaser. Førstevalget er lokal eksisjon, ellers er amputasjon en mulighet.
Behandling av lungemetastaser
Dersom man oppdager lungemetastaser bør pasienten vurderes for kirurgi. Ved et begrenset antall resektable metastaser er det en realistisk mulighet for kurasjon, både ved bensarkom og bløtvevssarkom. De viktigste prognostiske faktorene er antall metastaser, resektabilitet og tid siden primærbehandlingen. Kurasjon forutsetter som hovedregel at man kan gjøre pasienten makroskopisk tumorfri. Det er således ikke grunnlag for reseksjon av et begrenset antall metastaser i kurativt øyemed.
Strålebehandling
Sist faglig oppdatert: 28.02.2022
Palliativ strålebehandling brukes som tumorrettet og symptomlindrende behandling ved sarkom etter samme prinsipper som for andre kreftsykdommer, og har erfaringsmessig en betydelig plass. Standard fraksjonering er 3 Gy x 10-13 (Koontz, Clough, & Halperin, 2006). Det er mindre data vedrørende nytten av engangsfraksjoner ved skjelettmetastaser ved sarkom enn ved metastatisk karsinom, men engangsfraksjoner i form av 8 Gy x 1 kan benyttes, særlig der den kliniske behandlingsindikasjonen er smertelindring (Evidensgrad C/D).
Medikamentell behandling
Sist faglig oppdatert: 28.02.2022
Medikamentell behandling av metastatisk eller ikke-operabel lokalisert sykdom avhenger av sykdomsutbredelse og histologisk subtype og krever individuelle vurderinger. Behandlingen bør styres av sarkomsentere, og med lav terskel for diskusjon i multidisiplinære team.
Bløtvevssarkom
Ved begrenset metastatisk sykdom kan man legge kurativ målsetting på behandlingen, spesielt ved lungemetastaser. Om kjemoterapi har tilleggseffekt hos metastasektomerte pasienter er ikke avklart. Ved kjemoterapifølsomme svulster (f.eks. synovialt sarkom) og kurativ intensjon, er det i de fleste internasjonale miljøer vanlig å kombinere metastasektomi med kjemoterapi (Gronchi et al., 2021).
Ved metastatisk sykdom er kjemoterapiregimer basert på antrasykliner førstelinjes behandling (Evidensgrad A). Standard dosering er doksorubicin 75 mg/m2 gitt hver tredje uke. Lavere doser eller ukentlig doksorubicin kan vurderes ved nedsatt allmenntilstand eller komorbiditet. Det er ikke vist at kombinasjonsregimer gir bedre totaloverlevelse sammenlignet med doksorubicin monoterapi (Judson et al., 2014). Responsraten ved kombinasjonsbehandling er høyere, og kombinasjonen antrasyklin og ifosfamid bør vurderes ved histologiske undergrupper som er følsomme for ifosfamid, og dersom det anses fordelaktig å oppnå en dimensjonell respons (Evidensgrad A). Ved leiomyosarkom ser det ikke ut til å være noen gevinst av kombinasjonsbehandling med ifosfamid, men kombinasjonen doksorubicin og dakarbazin bør vurderes dersom objektiv respons er ønskelig å oppnå (D'Ambrosio et al., 2020) (Evidensgrad C). Toksisiteten ved kombinasjonskurene er signifikant høyere enn ved doksorubicin monoterapi, og dette bør vektlegges i vurderingen av behandlingsvalg (Judson et al., 2014).
Alternative regimer i andre og senere linjer er gemcitabin/docetaksel, trabektedin, pazopanib, eribulin, høydose ifosfamid, trofosfamid og dakarbazin. De ulike regimene er ikke sammenlignet direkte i prospektive studier, og noen klar anbefaling om prioritert rekkefølge kan ikke gis. Gemcitabin/docetaksel, trabektedin, pazopanib og eribulin har vist effekt i randomiserte fase III-studier og vil normalt være foretrukket. Mer utfyllende informasjon om de enkelte regimene følger under.
Kombinasjonen gemcitabin og docetaksel er vist å ha effekt ved flere histologiske undergrupper, spesielt ved leiomyosarkom, og kombinasjonsbehandling er mer effektivt enn gemcitabin alene (Evidensgrad B) (Hensley et al., 2002). Gemcitabin og docetaxel er sammenlignet med monoterapi doksorubicin i første linje i en randomisert studie som inkluderte de vanligste histologiske subtypene (Seddon et al., 2017). Det var ingen forskjell i progresjonsfri overlevelse eller totaloverlevelse, og heller ingen signifikant forskjell i toksisitet eller rapportert livskvalitet. Basert på tidligere ikke-randomiserte data har man antatt at pasienter med leiomyosarkomer har større nytte av gemcitabinbasert behandling, men det var i denne studien ingen signifikant forskjell mellom de histologiske undergruppene i subgruppeanalyser. Gemcitabin/docetaksel ansees som et alternativt førstelinjes regime (Evidensgrad A), men fordi doksorubicin er enklere å administrere (færre oppmøter og kortere tid), og billigere, vil det naturlige førstevalget fortsatt vanligvis være doksorubicin. Gemcitabin/docetaksel anbefales derfor hovedsakelig fra og med andre linje, alternativt i første linje i metastatisk situasjon, dersom antrasyklinbasert behandling er gitt adjuvant.
Trabektedin er sammenlignet med dakarbazin i en randomisert fase III-studie ved liposarkom og leiomyosarkom (G. D. Demetri et al., 2016). Det var en tydelig bedre progresjonsfri overlevelse i trabektedinarmen, men ingen signifikant forskjell i totaloverlevelse. En énarmet fase II-studie har vist at trabektedin kan gi sykdomskontroll også ved andre histologiske subtyper (Le Cesne et al., 2005). Responsraten ved myksoide liposarkomer er bedre enn ved øvrige subtyper, og kan tyde på at trabektedin bør foretrekkes som andrelinjes behandling ved denne undergruppen (Grosso et al., 2009). Medikamentet er lite benmargstoksisk, lite nyre- eller hjertetoksisk og gir sjelden hårtap. Alvorlig leverpåvirkning og rhabdomyolyse kan oppstå.
Pazopanib er en lite spesifikk tyrosinkinasehemmer som er registrert til behandling av voksne pasienter med spesifikke undergrupper av avansert bløtvevssarkom. Grunnlaget er data fra en fase II-studie med 142 pasienter (Sleijfer et al., 2009), og dernest en randomisert fase III-studie der pazopanib ble sammenlignet med placebo hos 372 pasienter med metastatisk, ikke-lipogent bløtvevssarkom. Det ble påvist en signifikant bedret median progresjonsfri overlevelse på 4,6 måneder mot 1,6 måneder i placebo-armen. Totaloverlevelse var imidlertid ikke signifikant bedre (van der Graaf et al., 2012) (Evidensgrad A). Anbefalt dose er 800 mg daglig. Medikamentet er et alternativ fra og med andre linje, men er ikke godkjent ved liposarkom. Bivirkningene er vanligvis moderate, men man må være oppmerksom på risiko for hypertensjon og påvirkning av leverfunksjon. Pazopanib er ikke behandlet i Nye Metoder.
Tubulinhemmeren eribulin er sammenlignet med dakarbazin i en randomisert fase III-studie hos pasienter med leiomyosarkom og liposarkom som tidligere har fått antracyklinbasert kjemoterapi (G. D. Demetri et al., 2017; Schöffski et al., 2016). Eribulin var assosiert med lenger progresjonsfri overlevelse og totaloverlevelse hos pasienter med liposarkom, men ikke leiomyosarkom. Medikamentet er basert på denne studien godkjent for pasienter med inoperabelt liposarkom som tidligere har fått antrasyklinholdig behandling. Eribulin ved inoperabelt/metastaserende liposarkom er vurdert i Nye Metoder. Beslutningsforum tok 30.08.21 følgende beslutning:
- Eribulin (Halaven) innføres til behandling av voksne pasienter med inoperabel liposarkom som tidligere har fått antracyklinholdig behandling (med mindre uegnet) mot avansert eller metastaserende sykdom.
- Det forutsetter at prisen er lik eller lavere enn den prisen som er grunnlaget for denne beslutningen.
- Behandlingen kan tas i bruk fra beslutningstidspunktet.
Eribulin anbefales som et alternativ fra og med andre linje ved liposarkom (Evidensgrad A). Det foreligger ikke data som støtter bruk av eribulin ved andre histologiske undergrupper.
Dakarbazin monoterapi kan ha effekt ved leiomyosarkom og solitær fibrøs tumor, og kan være et alternativ ved disse undergruppene (Stacchiotti et al., 2013; Van Glabbeke, Verweij, Judson, & Nielsen, 2002) (Evidensgrad C). Høydose ifosfamid er vist å gi objektiv respons, også etter behandling med standard dose ifosfamid, og kan være et alternativ, spesielt ved subtyper som er følsomme for ifosfamid (Le Cesne et al., 1995) (Evidensgrad C). Toksisiteten er imidlertid betydelig. Trofosfamid (Ixoten) er sammenlignet med monoterapi doksorubicin i en randomisert fase II-studie hos pasienter >60 år (Hartmann et al., 2020). Progresjonsfri overlevelse var sammenlignbar og toksisiteten var lavere med trofosfamid. Antrycyklinbasert behandling er aktuelt i andre linje, eller senere, dersom det ikke er gitt i første linje (Evidengrad B).
Histologisk subtype bør tillegges vekt i valg av medikamentell behandling fordi det er ulike responsrater ved ulike undergrupper. Det vil eksempelvis være mer aktuelt å gi kombinasjonsbehandling med ifosfamid ved synovialt sarkom og udifferensiert pleomorft sarkom fordi disse subtypene oppfattes å være mer følsomme for ifosfamid (Eriksson, 2010). Ved angiosarkomer er responsraten på behandling med taksaner i monoterapi høyere enn ved andre subgrupper, og monoterapi taksaner er et aktuelt alternativ fra og med andre linje (Evidensgrad B) (Penel et al., 2008). Antrasyklinbaserte regimer har begrenset effekt ved solitær fibrøs tumor og anbefales generelt ikke (Constantinidou et al., 2012). Enkelte sjeldne subtyper er ikke følsomme for kjemoterapi, som f.eks. alveolært bløtdelssarkom og klarcellet sarkom.
Det foreligger data som viser at immunsjekkpunkthemmere har effekt ved enkelte histologiske undergrupper (D'Angelo et al., 2018; Tawbi et al., 2017; Wilky et al., 2019), og at responsene kan være av lang varighet. SSG har publisert anbefalinger for behandling med immunsjekkpunkthemmere ved sarkom, og det vises til disse (ssg-org.net). Ingen immunsjekkpunkthemmere har markedsføringstillatelse for sarkom, og de er heller ikke vurdert i Nye Metoder.
Nytten man kan forvente å oppnå ved medikamentell behandling hos pasienter med utbredte metastaser kan ofte være begrenset. Det er viktig å gi realistisk informasjon til pasienten og å balansere potensiell nytte mot risiko for bivirkninger. I enkelte tilfeller vil det være riktig å ikke tilby videre systemisk tumorrettet behandling. Smertelindrende medikamentell behandling, strålebehandling og annen symptomrettet behandling vil ofte være viktigere å tilby pasientene i sene sykdomsfaser.
Anbefalinger:
- Ved metastatisk sykdom er kjemoterapiregimer basert på antrasykliner førstelinjes behandling (Evidensgrad A).
- Aktuelle regimer i andre og senere linjer er gemcitabin/docetaksel (Evidensgrad A), trabektedin (Evidensgrad A), pazopanib (Evidensgrad A), eribulin (Evidensgrad A), høydose ifosfamid (Evidensgrad B), trofosfamid (Evidensgrad A) og dakarbazin (Evidensgrad A).
- Histologisk subtype bør tillegges vekt i valg av regime fordi det er ulike responsrater ved enkelte undergrupper (Evidensgrad C).
Gastrointestinal stromal tumor
Førstelinjes behandling ved metastatisk eller lokalisert, ikke-operabel GIST er tyrosinkinashemmeren imatinib (Evidensgrad A). Mutasjonsanalyse skal utføres i forbindelse med oppstart av behandling. Over 90% av pasientene med svulster med mutasjon i KIT eller PDGFRA har effekt av imatinib. Anbefalt dose imatinib er 400 mg x 1. Ved mutasjon i KIT ekson 9 anbefales det å forsøke doseøkning til 800 mg x 1 fordi økt dose i denne undergruppen ser ut til å gi bedret progresjonsfri overlevelse (Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST), 2010). Det foreligger ikke studier som viser at konsentrasjonsmåling av imatinib i plasma eller serum bør være klinisk rutine, men det kan muligens være fordelaktig i spesielle situasjoner, som f.eks. ved uvanlig kraftige bivirkninger med tanke på å styre dosereduksjon. Bivirkningene av imatinib er som regel moderate, og ødemer, kvalme, diaré, dermatitt og fatigue er de vanligste.
Behandling med imatinib bør gis livslangt eller til progresjon. Seponering er vist i en randomisert studie å føre til progresjon hos nesten alle pasientene (Le Cesne et al., 2010). En skandinavisk studie har undersøkt om seponering kan være trygt hos pasienter med oligometastatisk sykdom etter minimum 5 års behandling med imatinib og lokalbehandling av alle metastaser (SSG XXV: The Stop-GIST Trial; Discontinuation of Imatinib in Patients With Oligo-metastatic GIST, 2016-2022). Studien er stengt for inklusjon, men resultatene foreligger foreløpig ikke.
Svulster uten påvist mutasjon i KIT eller PDGFRA responderer vanligvis ikke på imatinib. Likevel anbefales ofte imatinib i fravær av andre behandlingsalternativer, og fordi mutasjonsanalysen i enkelte tilfeller kan være falsk negativ. Svulster med D842V mutasjon i PDGFRA genet responderer ikke på imatinib. Avapritinib er en ny tyrosinkinasehemmer med meget god effekt ved D842V-mutert GIST (Jones et al., 2021), og er godkjent av EMA, og er til vurdering i Nye Metoder.
Ved progresjon på imatinib kan lokalbehandling i form av kirurgi, RFA eller strålebehandling vurderes dersom det er lokal og begrenset progresjon, ofte vurdert som inntil tre progredierende metastaser. Dersom det er behov for å endre medikamentell behandling er doseøkning av imatinib til 800 mg x 1 et alternativ (Zalcberg et al., 2005), spesielt dersom pasienten har lite bivirkninger ved standard dose. Anbefalt andrelinjes behandling er sunitinib gitt i 6 ukers syklus med 4 uker i dosering 50 mg x 1 etterfulgt av 2 ukers pause (G.D. Demetri et al., 2006) (Evidensgrad A). Anbefalt tredjelinjes behandling er regorafenib (G. D. Demetri et al., 2013) (Evidensgrad A). Både sunitinib og regorafenib gir en median gevinst i progresjonsfri overlevelse på 4-5 måneder sammenlignet med placebo. Begge medikamenter kan gi betydelige bivirkninger, og en avveining av nytteverdi mot bivirkninger bør gjøres fortløpende under behandlingen.
Andre tyrosinkinasehemmere er også vist å ha effekt ved GIST. Disse inkluderer blant annet ripretinib, pazopanib, nilotinib og sorafenib (Blay et al., 2020; Ferraro & Zalcberg, 2014; Garcia del Muro, 2015; Joensuu et al., 2015; Mir et al., 2016). Ingen av disse er foreløpig vurdert i Nye Metoder. Det er vist at fortsatt behandling med en tyrosinkinasehemmer etter progresjon kan forsinke ytterligere sykdomsutvikling, og at behandling med imatinib i senere linjer kan gi gevinst tross tidligere progresjon på imatinib (Kang et al., 2013). Å kontinuere behandling tross progresjon, eller reoppstart imatinib, er således et alternativ.
Anbefalinger:
- Imatinib anbefales som førstelinjes behandling ved metastatisk eller lokalisert, ikke-operabel GIST (Evidensgrad A).
- Behandling med imatinib bør gis livslangt eller til progresjon (Evidensgrad A).
- Anbefalt andrelinjes behandling er sunitinib (Evidensgrad A).
- Anbefalt tredjelinjes behandling er regorafenib (Evidensgrad A).
Osteosarkom
Ved metastatisk sykdom på diagnosetidspunkt kan det legges kurativ målsetting på behandlingen dersom alle metastaser er resektable (S.S. Bielack & Carrle, 2008) (Evidensgrad B). I praksis betyr dette pasienter med begrenset spredning til lungene. Langtidsoverlevelse opp mot 40 % har vært rapportert ved kjemoterapi kombinert med kirurgi. Kjemoterapi gis etter samme anbefalinger som for lokalisert sykdom (Evidensgrad A). Kjemoterapi med perifer stamcellestøtte (HMAS) har vært utprøvd for pasienter med metastaser på diagnosetidspunktet (protokoll ISG/SSG II) (An italian - scandinavian treatment protocol for metastatic and pelvic osteosarcoma (ISG/SSG II), 2002), men resultatene var ikke bedre enn ved konvensjonell behandling (Boye et al., 2014).
Ved tilbakefall er kurativ målsetting også mulig dersom all sykdom er resektabel. Elementer som må vektlegges i valg av behandling er antall metastaser, lokalisasjon av metastaser og tid etter primærbehandlingen. Kirurgi er som hovedregel eneste behandling ved få og resektable lungemetastaser, spesielt hvis tid siden primærbehandling er lang. Betydningen av kjemoterapi ved tilbakefall er ikke avklart, men flere rapporter tyder på en viss effekt. Dersom kurativt siktemål kan legges til grunn, anbefales tillegg av høydose ifosfamid, eventuelt i kombinasjon med etoposid, for pasienter som tidligere ikke har fått slik behandling (S.S. Bielack & Carrle, 2008; Strauss et al., 2021) (Evidensgrad B). Aktuelle regimer ved livsforlengende behandling er i tillegg til høydose ifosfamid (med eller uten etoposid), gemcitabin/docetaksel (Palmerini et al., 2016), karboplatin/etoposid (Van Winkle et al., 2005), regorafenib (L. E. Davis et al., 2019; Duffaud et al., 2019) og pazopanib (Longhi et al., 2019). Regorafenib og pazopanib har ikke vært til vurdering i Nye Metoder.
Anbefalinger:
- Ved metastatisk sykdom på diagnosetidspunkt kan det legges kurativ målsetting på behandlingen dersom alle metastaser er resektable (Evidensgrad D).
- Kjemoterapi i første linje gis etter samme anbefalinger som for lokalisert sykdom, i henhold til EURAMOS-1 protokollen <40 år (Evidensgrad A) og i henhold til EURO-B.O.S.S.-protokollen >40 år (Evidensgrad B).
- Aktuelle regimer i andre og senere linjer er høydose ifosfamid med eller uten etoposid (Evidensgrad B), gemcitabin/docetaksel (Evidensgrad C), karboplatin/etoposid (Evidensgrad B), regorafenib (Evidensgrad A) og pazopanib (Evidensgrad C).
Ewing sarkom
Ved metastatisk sykdom kun til lunge og/eller pleura på diagnosetidspunkt er det utført to studier (Euro-E.W.I.N.G. 99 og Ewing 2008) som har sammenlignet konvensjonell behandling med HMAS, og resultatene fra disse er analysert samlet (Dirksen et al., 2019). Alle pasienter fikk induksjonsbehandling med VIDE, og ble randomisert til 1 VAI + HMAS eller 8 VAI + total lungebestråling som konsoliderende behandling. Det var en ikke-signifikant forskjell til fordel for HMAS når det gjaldt hendelsesfri overlevelse («event-free survival»), men ingen forskjell i totaloverlevelse. Akutt toksisitet var som forventet høyere med HMAS. I ISG/SSG IV-studien som hovedsakelig inkluderte pasienter med lungemetastaser ble det gitt både HMAS og total lungebestråling (Luksch et al., 2012). Studien var ikke randomisert, og således vanskelig å sammenligne direkte med andre studier, men resultatene er ikke åpenbart bedre enn med konvensjonell kjemoterapi og total lungebestråling. Basert på de foreliggende studiene anbefaler de fleste internasjonale grupper ikke HMAS til pasienter med metastaser til lunge/pleura, og man bør som hovedregel følge disse internasjonale anbefalingene. Det anbefales således behandling i henhold til Euro Ewing 2012-protokollen uten HMAS (Evidensgrad A).
Ved metastaser utenfor lunge/pleura på diagnosetidspunkt er det ingen studier som entydig kan avgjøre hva som er den beste behandlingen, og beslutningen vil til en viss grad bli en individuell vurdering avhengig av sykdomsutbredelse, risikofaktorer, tilgjengelighet for adekvat lokal behandling og eventuelle kontraindikasjoner mot HMAS (Ladenstein et al., 2010; Oberlin et al., 2006). Anbefalt induksjonsbehandling i første linje er VDC/IE i henhold til Euro Ewing 2012-protokollen (Evidensgrad A). Som konsoliderende behandling er det to alternativer; VC/IE som ved lokalisert sykdom eller HMAS. Upubliserte og preliminære data fra Ewing 2008-studien viser at HMAS med treosulfan og melfalan ved metastatisk sykdom gir bedre langtidsoverlevelse hos pasienter <14 år (Uta Dirksen, personlig kommunikasjon). I denne protokollen var VIDE gitt som induksjonsbehandling. Foreløpige data fra Euro Ewing 2012 viser at VDC/IE er bedre enn VIDE også i denne pasientgruppen, og det er således uklart om resultatene vil være overførbare. HMAS kan vurderes hos denne pasientgruppen etter en individuell vurdering, og etter diskusjon om fordeler og ulemper med pasient/foreldre.
Ved tilbakefall etter, eller progresjon underveis i primærbehandlingen, er prognosen dårlig. Ved begrenset sykdomsutbredelse, spesielt ved isolert lokalt residiv eller ved isolerte lungemetastaser, er kurativt siktemål mulig. Høydose ifosfamid anbefales som andre linjes behandling ved kurativt siktemål. Lokalbehandling av all makroskopisk sykdom er en forutsetning. Kirurgi og/eller strålebehanding er aktuelle modaliteter.
Ved ikke-resektabel, utbredt sykdom er kurativt siktemål urealistisk. Det foreligger ikke publiserte studier som har sammenlignet ulike regimer. Den pågående, internasjonale fase 2/3 studien rEECur har som mål å sammenligne ulike regimer ved residiverende eller primært refraktær Ewing sarkom, og det anbefales inklusjon i denne studien hvis mulig. Inkluderte regimer så langt er høydose ifosfamid, cyklofosfamid/topotekan, gemcitabin/docetaksel, irinotekan/temozolomid og karboplatin/etoposid. Foreløpige data har vist at høydose ifosfamid ser ut til å være mest effektivt, men endelige resultater fra studien foreligger ikke. Dersom inklusjon i studien ikke er mulig, oppfattes alle de ovennevnte regimene som aktuelle alternativer.
Anbefalinger:
- Ved metastatisk sykdom kun til lunge og/eller pleura på diagnosetidspunkt anbefales behandling i henhold til Euro Ewing 2012-protokollen med total lungebestråling uten HMAS (Evidensgrad A).
- Ved metastaser utenfor lunge/pleura på diagnosetidspunkt anbefales behandling i henhold til Euro Ewing 2012-protokollen (Evidensgrad A). HMAS kan vurderes etter en individuell vurdering, og etter diskusjon om fordeler og ulemper med pasient/foreldre (Evidensgrad B).
- Ved primært refraktær sykdom eller residiv etter primærbehandling, anbefales inklusjon i rEECur-studien eller behandling i henhold til denne (Evidensgrad D).
Metode og prosess for utarbeidelse av retningslinjene
Hva er nasjonale retningslinjer?
Sist faglig oppdatert: 28.02.2022
Nasjonal helse- og sykehusplan (2016–2019) (Helse- og omsorgsdepartementet, 2015) klargjør at Helsedirektoratet innenfor rettslige rammer, har en normerende rolle for helsetjenesten på tvers av helseregioner og tjenestenivå. Helsedirektoratet er derved eneste aktør som har mandat til å lage nasjonale retningslinjer for helsetjenesten. Helsedirektoratet har en koordinerende rolle for å utvikle overordnede referanserammer for kreftomsorgen, sammen med de regionale helseforetakene, kommunene og andre relevante myndighetsorganer og tjenester.
Nasjonale retningslinjer fra Helsedirektoratet er å betrakte som anbefalinger og råd, basert på oppdatert faglig kunnskap som er fremskaffet på en systematisk, kunnskapsbasert måte. De nasjonale retningslinjene gir uttrykk for hva som anses som god praksis på utgivelsestidspunktet, og er ment som et hjelpemiddel ved de avveininger tjenesteyterne må gjøre for å oppnå forsvarlighet og god kvalitet i tjenesten.
Nasjonale retningslinjer er ikke rettslig bindende for mottakerne, men skal som faglig normerende langt på vei være styrende for de valg som skal tas. Ved å følge oppdaterte nasjonale retningslinjer vil fagpersonell bidra til å oppfylle kravet om faglig forsvarlighet i lovverket. Dersom en velger løsninger som i vesentlig grad avviker fra nasjonale faglige retningslinjer, bør en dokumentere dette og være forberedt til å begrunne sitt valg.
Kunnskapsbasert prosess
Sist faglig oppdatert: 28.02.2022
Helsedirektoratet legger til grunn at alle nasjonale retningslinjer skal være utarbeidet etter en metode med vekt på forskningsbasert kunnskap, tydelig og tilgjengelig dokumentasjon, brukermedvirkning, tverrfaglighet, fokus på praksis, implementering og oppdatering.
Det er en omfattende prosess å lage gode retningslinjer som tilfredsstiller krav til prosess, metode og transparens, som er det nivået Helsedirektoratet og andre internasjonale organisasjoner har lagt til grunn for utforming av anbefalinger.
Gradering av kunnskapsgrunnlaget
Sist faglig oppdatert: 28.02.2022
Ved utarbeiding av nasjonale retningslinjer stilles det krav om at all relevant kunnskap på området hentes frem, beskrives og vurderes på en systematisk og åpen måte.
I disse retningslinjene har man benyttet følgende graderingsmodell for å vise hvilket vitenskapelig grunnlag kunnskapen er basert på.
| Studietype | Evidensnivå | Gradering av evidensnivå |
|---|---|---|
| Kunnskap som bygger på systematiske oversikter og meta‑analyser av randomiserte kontrollerte studier | Nivå 1a | A |
| Kunnskap som bygger på minst én randomisert kontrollert studie | Nivå 1b |
|
| Kunnskap som bygger på minst én godt utformet kontrollert studie uten randomisering | Nivå 2a | B |
| Kunnskap som bygger på minst én annen godt utformet kvasi-eksperimentell studie uten randomisering | Nivå 2b |
|
| Kunnskap som bygger på godt utformede ikke eksperimentelle beskrivende studier, som sammenlignende studier, korrelasjonsstudier og case studier | Nivå 3 | C |
| Kunnskap som bygger på rapporter eller oppfatninger fra eksperter, komiteer og/eller klinisk ekspertise hos respekterte autoriteter | Nivå 4 | D |
I disse retningslinjene er kun kunnskapsgrunnlaget og ikke anbefalingene gradert. Hvis selve anbefalingene skal graderes må man, i tillegg til å ha vurdert kunnskapsgrunnlaget, også legge inn en vurdering av både kost-nytte og andre forhold (klinisk erfaring, skjønn, etikk, osv). Dette er ikke gjort eksplisitt i forbindelse med dette arbeidet, og anbefalingene er derfor ikke gradert.
For å indikere kunnskapsgrunnlaget for anbefalingene, er det brukt gradering A–D. En anbefaling som er anført med D behøver derfor ikke å være en svakere anbefaling enn en som er anført A, B eller C. Det hen speiler kun til kunnskapsgrunnlaget anbefalingen er basert på. I enkelte sammenhenger vil klinisk erfaring og gjeldende praksis være et godt grunnlag for anbefalingen. I andre sammenhenger er det uttrykk for at det ikke finnes tilstrekkelig vitenskapelig dokumentasjon for anbefalingen, selv om det hadde vært ønskelig.
Nasjonalt handlingsprogram – 1. utgave (6/2012 – IS-1980)
Sist faglig oppdatert: 28.02.2022
Den første utgaven av nasjonale retningslinjer for sarkom ble skrevet av en arbeidsgruppe med representanter fra Norsk sarkomgruppe, samt andre relevante fagpersoner. Arbeidsgruppen besto av følgende personer:
- Sigmund Skjeldal (ortoped), Kirurgisk klinikk, Radiumhospitalet
- Kirsten Sundby Hall (onkolog), Onkologisk avd., Radiumhospitalet
- Ingeborg Taksdal (radiolog), Avd for radiologi og nukleærmedisin, Klinikk for diagnostikk og intervensjon, Oslo Universitetsklinikk, Radiumhospitalet.
- Ayca M. Løndalen, Nukleærmedisinsk avd, Radiumhospitalet
- Bodil Bjerkehagen (patolog), Patologiklinikken, Radiumhospitalet
- Stephan Stoldt, Kirurgisk avdeling, Radiumhospitalet
- Olga Zaikova (ortoped) Kirurgisk klinikk, Radiumhospitalet
- Ole-Jacob Norum (ortoped), Kirurgisk klinikk, Radiumhospitalet.
- Gunnar B. Kristensen (gynekolog), Avd for gynekologisk kreft, Radiumhospitalet
- Heidi Glosli (barneonkolog), Barneklinikken, Rikshospitalet
- Annja Viset (radiolog), Klinikk for Bildediagnostikk, Seksjon for Muskel-/skjelett radiologi, St. Olavs hospital
- Clement Trovik (ortoped), Kreftavdelingen/Ortopedisk avd, Haukeland universitetssykehus
- Svein Halvorsen (radiolog), Radiologisk avd., Haukeland universitetssykehus
- Eivind Smeland (onkolog), Kreftavd., Universitetssykehuset Nord-Norge
- Trond Viset (patolog), Avd. for Patologi og Medisinsk Genetikk, St. Olavs hospital
- Odd Monge (onkolog), Senter for ben- og bløtvevssvulster, Haukeland universitetssykehus
- Nina Jebsen (onkolog), Kreftklinikken, Haukeland Universitetssykehus
- Hans Kristian Haugland (patolog), Avd. for Patologi, Haukeland universitetssykehus
- Heidi Knobel (onkolog), Kreftavdelingen, St. Olavs hospital
- Vidar Isaksen (patolog), Patologisk anatomisk avd., Universitetssykehuset Nord-Norge
- Charlott Maria Våde (sykepleier), Radiumhospitalet
- Synnøve Granlien (sykepleier), Radiumhospitalet
- Anicken Smith Huun (fysioterapeut), Fysioterapiavdelingen, Haukeland Universitetssykehus
- Merethe Lia Johansen (fysioterapeut), Klinikk for klinisk service, Radiumhospitalet
- Marit Gudim (sosionom), Radiumhospitalet
- Hege Thorsrud (klinisk ernæringsfysiolog), Radiumhospitalet
- Trine Thoresen (sekretær), Kompetansesenter for sarkom, Radiumhospitalet
- Lene Juvet (forsker), Nasjonalt kunnskapssenter for helsetjenesten
Nasjonalt handlingsprogram – 2. utgave (3/2015 – IS-2218)
Sist faglig oppdatert: 28.02.2022
Retningslinjen fra 2012 ble gjennomgått og oppdatert av samme arbeidsgruppe, og ny oppdatert utgave forelå i mars 2015.
Nasjonalt handlingsprogram – 3. utgave (4/2018 – IS-2697)
Sist faglig oppdatert: 28.02.2022
Utgaven fra 2015 ble gjennomgått og oppdatert (2017/2018) av følgende fagpersoner:
- Olga Zaikova (leder) – ortoped
- Kjetil Boye – onkolog
- Kirsten Sundby Hall – onkolog
- Ingeborg Taksdal – radiolog
- Bodil Bjerkehagen – patolog
- Nina Jebsen – onkolog
- Toto Hølmebakk – gastrokirurg
- Ayca M. Løndalen – nukleærmedisiner
- Heidi Glosli – barneonkolog
- Tone Skeie-Jensen – gynekologisk sarkom
I tillegg var det fagpersoner som ga faglige bidrag til oppdateringen:
Fra OUS
- Stephan Stoldt – gastrokirurg
- Jan Peter Poulsen – onkolog
- Øyvind Bruland – onkolog
- Charlott Maria Våde – sykepleier
- Merethe Lia Johansen – fysioterapeut
- Marit Gudim – sosionom
- Hege Thorsrud – Klinisk ernæringsfysiolog
- Trine Thoresen – sekretær
Fra Haukeland universitetssykehus
- Hans Kristian Haugland – patolog
- Clement Trovik – ortoped
- Svein Halvorsen – radiolog
Fra St Olavs hospital
- Heidi Knobel – onkolog
- Trond Viset – patolog
- Annja Viset – radiolog
Fra Universitetssykehuset i Nord-Norge
- Vidar Isaksen – patolog
- Eivind Smeland – onkolog
Nasjonalt handlingsprogram – 4. utgave (2/2022 – IS-3028)
Sist faglig oppdatert: 28.02.2022
Utgaven fra 2018 er revidert av følgende arbeidsgruppe oppnevnt av Helsedirektoratet:
- Kjetil Boye (leder) – onkolog
- Heidi Knobel – onkolog
- Annja Viset – radiolog
- Thomas Kilvær – onkolog
- Olga Zaikova – ortoped
- Kirsten Sundby Hall – onkolog
- Bodil Bjerkehagen – patolog
- Toto Hølmebakk – gastrokirurg
- Ayca M. Løndalen – nukleærmedisiner
- Heidi Glosli – barneonkolog
- Tone Skeie-Jensen – gynekolog
- Hans Kristian Haugland – patolog
- Nina L. Jebsen – onkolog
- Anders Sund – ortoped
- Karin Frydenberg – fastlege
I tillegg ga følgende fagpersoner faglige bidrag til oppdateringen:
- Øyvind Bruland – onkolog
- Ingeborg Taksdal – radiolog
Referanser
Sist faglig oppdatert: 28.02.2022
Abeler, V. M., Røyne, O., Thoresen, S., Danielsen, H. E., Nesland, J. M., & Kristensen, G. B. (2009). Uterine sarcomas in Norway. A histopathological and prognostic survey of a total population from 1970 to 2000 including 419 patients. Histopathology, 54(3), 355-364.
Aisen, A. M., Martel, W., Braunstein, E. M., McMillin, K. I., Phillips, W. A., & Kling, T. F. (1986). MRI and CT evaluation of primary bone and soft-tissue tumors. AJR Am J Roentgenol, 146(4), 749-756.
Aksnes, L. H. (2008). Health in long-term survivors of bone sarcoma (Doktorgrad). Oslo: Faculty of Medicine, University of Oslo Unipub.
Al-Absi, E., Farrokhyar, F., Sharma, R., Whelan, K., Corbett, T., Patel, M., & Ghert, M. (2010). A systematic review and meta-analysis of oncologic outcomes of pre- versus postoperative radiation in localized resectable soft-tissue sarcoma. Ann Surg Oncol, 17(5), 1367-1374.
Albertsmeier, M., Rauch, A., Roeder, F., Hasenhütl, S., Pratschke, S., Kirschneck, M., . . . Angele, M. K. (2018). External Beam Radiation Therapy for Resectable Soft Tissue Sarcoma: A Systematic Review and Meta-Analysis. Ann Surg Oncol, 25(3), 754-767. https://doi.org/10.1245/s10434-017-6081-2
Alektiar, K. M., Brennan, M. F., Healey, J. H., & Singer, S. (2008). Impact of intensity-modulated radiation therapy on local control in primary soft-tissue sarcoma of the extremity. J Clin Oncol, 26(20), 3440-3444.
Alektiar, K. M., Velasco, J., Zelefsky, M. J., Woodruff, J. M., Lewis, J. J., & Brennan, M. F. (2000). Adjuvant radiotherapy for margin-positive high-grade soft tissue sarcoma of the extremity. Int J Radiat Oncol Biol Phys, 48(4), 1051-1058.
Allen, A. M., Pawlicki, T., Dong, L., Fourkal, E., Buyyounouski, M., Cengel, K., . . . Konski, A. A. (2012). An evidence based review of proton beam therapy: the report of ASTRO's emerging technology committee. Radiother Oncol, 103(1), 8-11. https://doi.org/10.1016/j.radonc.2012.02.001
Alyas, F., James, S. L., Davies, A. M., & Saifuddin, A. (2007). The role of MR imaging in the diagnostic characterisation of appendicular bone tumours and tumour-like conditions. Eur Radiol, 17(10), 2675-2686.
Andreassen, A., Øyjord, T., Hovig, E., Holm, R., Flørenes, V. A., Nesland, J. M., . . . Børresen, A. L. (1993). p53 abnormalities in different subtypes of human sarcomas. Cancer Research, 53(3), 468-471.
Arends, J., Bachmann, P., Baracos, V., Barthelemy, N., Bertz, H., Bozzetti, F., . . . Preiser, J. C. (2017). ESPEN guidelines on nutrition in cancer patients. Clin Nutr, 36(1), 11-48. https://doi.org/10.1016/j.clnu.2016.07.015
Arlen, M., Higinbotham, N. L., Huvos, A. G., Marcove, R. C., Miller, T., & Shah, I. C. (1971). Radiation-induced sarcoma of bone. Cancer, 28(5), 1087-1099.
Bauer, H. C., Trovik, C. S., Alvegard, T. A., Berlin, O., Erlanson, M., Gustafson, P., . . . Wiklund, T. (2001). Monitoring referral and treatment in soft tissue sarcoma: study based on 1,851 patients from the Scandinavian Sarcoma Group Register. Acta Orthop Scand, 72(2), 150-159.
Becher, S., & Oskouei, S. (2015). PET Imaging in Sarcoma. Orthopedic Clinics of North America, 46(3), 409-415, xi. https://doi.org/10.1016/j.ocl.2015.03.001
Berger-Richardson, D., & Swallow, C. J. (2017). Needle tract seeding after percutaneous biopsy of sarcoma: Risk/benefit considerations. Cancer, 123(4), 560-567. https://doi.org/10.1002/cncr.30370
Berner, K., Johannesen, T. B., Berner, A., Haugland, H. K., Bjerkehagen, B., Bøhler, P. J., & Bruland, O. S. (2015). Time-trends on incidence and survival in a nationwide and unselected cohort of patients with skeletal osteosarcoma. Acta Oncologica, 54(1), 25-33.
Berquist, T. H., Ehman, R. L., King, B. F., Hodgman, C. G., & Ilstrup, D. M. (1990). Value of MR imaging in differentiating benign from malignant soft-tissue masses: study of 95 lesions. AJR Am J Roentgenol, 155(6), 1251-1255.
Bhangu, A. A., Beard, J. A., & Grimer, R. J. (2004). Should soft tissue sarcomas be treated at a specialist centre? Sarcoma, 8(1), 1-6.
Bielack, S. S., & Carrle, D. (2008). State-of-the-art approach in selective curable tumors: bone sarcoma. Ann Oncol, 19 Suppl 7, vii155-vii160.
Bielack, S. S., Smeland, S., Whelan, J. S., Marina, N., Jovic, G., Hook, J. M., . . . investigators, E.-. (2015). Methotrexate, Doxorubicin, and Cisplatin (MAP) Plus Maintenance Pegylated Interferon Alfa-2b Versus MAP Alone in Patients With Resectable High-Grade Osteosarcoma and Good Histologic Response to Preoperative MAP: First Results of the EURAMOS-1 Good Response Randomized Controlled Trial. Journal of Clinical Oncology, 33(20), 2279-2287.
Bischof, D. A., Kim, Y., Dodson, R., Jimenez, M. C., Behman, R., Cocieru, A., . . . Pawlik, T. M. (2015). Conditional disease-free survival after surgical resection of gastrointestinal stromal tumors: a multi-institutional analysis of 502 patients. JAMA Surg, 150(4), 299-306. https://doi.org/10.1001/jamasurg.2014.2881
Bjerkehagen, B., Smeland, S., Walberg, L., Skjeldal, S., Hall, K. S., Nesland, J. M., . . . Saeter, G. (2008). Radiation-induced sarcoma: 25-year experience from the Norwegian Radium Hospital. Acta oncologica (Stockholm, Sweden), 47(8), 1475-1482.
Blanchard, D. K., Reynolds, C., Grant, C. S., Farley, D. R., & Donohue, J. H. (2002). Radiation-induced breast sarcoma. Am J Surg, 184(4), 356-358.
Blay, J. Y., Serrano, C., Heinrich, M. C., Zalcberg, J., Bauer, S., Gelderblom, H., . . . von Mehren, M. (2020). Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. The Lancet. Oncology, 21(7), 923-934. https://doi.org/10.1016/s1470-2045(20)30168-6
Bonvalot, S., Gronchi, A., Le Péchoux, C., Swallow, C. J., Strauss, D., Meeus, P., . . . Haas, R. L. (2020). Preoperative radiotherapy plus surgery versus surgery alone for patients with primary retroperitoneal sarcoma (EORTC-62092: STRASS): a multicentre, open-label, randomised, phase 3 trial. The Lancet. Oncology, 21(10), 1366-1377. https://doi.org/10.1016/s1470-2045(20)30446-0
Bonvalot, S., Rivoire, M., Castaing, M., Stoeckle, E., Le Cesne, A., Blay, J. Y., & Laplanche, A. (2009). Primary retroperitoneal sarcomas: a multivariate analysis of surgical factors associated with local control. J Clin Oncol, 27(1), 31-37. https://doi.org/10.1200/jco.2008.18.0802
Bosma, S. E., Cleven, A. H. G., & Dijkstra, P. D. S. (2019). Can Navigation Improve the Ability to Achieve Tumor-free Margins in Pelvic and Sacral Primary Bone Sarcoma Resections? A Historically Controlled Study. Clin Orthop Relat Res, 477(7), 1548-1559. https://doi.org/10.1097/corr.0000000000000766
Boye, K., Del Prever, A. B., Eriksson, M., Saeter, G., Tienghi, A., Lindholm, P., . . . Hall, K. S. (2014). High-dose chemotherapy with stem cell rescue in the primary treatment of metastatic and pelvic osteosarcoma: final results of the ISG/SSG II study. Pediatric blood & cancer, 61(5), 840-845. https://doi.org/10.1002/pbc.24868
Brennan, M. F., Antonescu, C. R., Moraco, N., & Singer, S. (2014). Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg, 260(3), 416-421; discussion 421-412. https://doi.org/10.1097/sla.0000000000000869
Brenner, D. J., Curtis, R. E., Hall, E. J., & Ron, E. (2000). Second malignancies in prostate carcinoma patients after radiotherapy compared with surgery. Cancer, 88(2), 398-406.
Cahan, W. G., Woodard, H. Q., Higinbotham, N. L., Stewart, F. W., & Coley, B. L. (1998). Sarcoma arising in irradiated bone: report of eleven cases. 1948. Cancer, 82(1), 8-34.
Canter, R. J., Qin, L. X., Ferrone, C. R., Maki, R. G., Singer, S., & Brennan, M. F. (2008). Why do patients with low-grade soft tissue sarcoma die? Ann Surg Oncol, 15(12), 3550-3560.
Casali, P. G., Blay, J. Y., Abecassis, N., Bajpai, J., Bauer, S., Biagini, R., . . . Stacchiotti, S. (2022). Gastrointestinal stromal tumours: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol, 33(1), 20-33. https://doi.org/10.1016/j.annonc.2021.09.005
Cha, C., Antonescu, C. R., Quan, M. L., Maru, S., & Brennan, M. F. (2004). Long-term results with resection of radiation-induced soft tissue sarcomas. Ann Surg, 239(6), 903-909.
Chen, L., Wu, X., Ma, X., Guo, L., Zhu, C., & Li, Q. (2017). Prognostic value of 18F-FDG PET-CT-based functional parameters in patients with soft tissue sarcoma: A meta-analysis. Medicine (Baltimore), 96(6), e5913. https://doi.org/10.1097/md.0000000000005913
Chouliaras, K., Senehi, R., Ethun, C. G., Poultsides, G., Grignol, V., Clarke, C. N., . . . Votanopoulos, K. I. (2019). Role of radiation therapy for retroperitoneal sarcomas: An eight-institution study from the US Sarcoma Collaborative. Journal of Surgical Oncology, 120(7), 1227-1234. https://doi.org/10.1002/jso.25694
Ciernik, I. F., Niemierko, A., Harmon, D. C., Kobayashi, W., Chen, Y. L., Yock, T. I., . . . Delaney, T. F. (2011). Proton-based radiotherapy for unresectable or incompletely resected osteosarcoma. Cancer, 117(19), 4522-4530. https://doi.org/10.1002/cncr.26037
College of American Pathologists. (2012). [nettside]. Northfield, IL: CAP. Hentet 2012, fra www.cap.org/
Constantinidou, A., Jones, R. L., Olmos, D., Thway, K., Fisher, C., Al-Muderis, O., & Judson, I. (2012). Conventional anthracycline-based chemotherapy has limited efficacy in solitary fibrous tumour. Acta oncologica (Stockholm, Sweden), 51(4), 550-554. https://doi.org/10.3109/0284186x.2011.626450
Cramp, F., & Daniel, J. (2008). Exercise for the management of cancer-related fatigue in adults. Cochrane Database Syst Rev, (2), CD006145.
D'Ambrosio, L., Touati, N., Blay, J. Y., Grignani, G., Flippot, R., Czarnecka, A. M., . . . Gronchi, A. (2020). Doxorubicin plus dacarbazine, doxorubicin plus ifosfamide, or doxorubicin alone as a first-line treatment for advanced leiomyosarcoma: A propensity score matching analysis from the European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. Cancer, 126(11), 2637-2647. https://doi.org/10.1002/cncr.32795
D'Angelo, S. P., Mahoney, M. R., Van Tine, B. A., Atkins, J., Milhem, M. M., Jahagirdar, B. N., . . . Streicher, H. (2018). Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. The Lancet. Oncology, 19(3), 416-426. https://doi.org/10.1016/s1470-2045(18)30006-8
Daisne, J. F., Duprez, T., Weynand, B., Lonneux, M., Hamoir, M., Reychler, H., & Gregoire, V. (2004). Tumor volume in pharyngolaryngeal squamous cell carcinoma: comparison at CT, MR imaging, and FDG PET and validation with surgical specimen. Radiology, 233(1), 93-100.
Dangoor, A., Seddon, B., Gerrand, C., Grimer, R., Whelan, J., & Judson, I. (2016). UK guidelines for the management of soft tissue sarcomas. Clinical sarcoma research, 6, 20. https://doi.org/10.1186/s13569-016-0060-4
Davis, A. M., O'Sullivan, B., Turcotte, R., Bell, R., Catton, C., Chabot, P., . . . Trial, N. C. I. C. C. T. G. R. (2005). Late radiation morbidity following randomization to preoperative versus postoperative radiotherapy in extremity soft tissue sarcoma. Radiotherapy and Oncology, 75(1), 48-53.
Davis, L. E., Bolejack, V., Ryan, C. W., Ganjoo, K. N., Loggers, E. T., Chawla, S., . . . Maki, R. G. (2019). Randomized Double-Blind Phase II Study of Regorafenib in Patients With Metastatic Osteosarcoma. J Clin Oncol, 37(16), 1424-1431. https://doi.org/10.1200/jco.18.02374
DeMatteo, R. P., Lewis, J. J., Leung, D., Mudan, S. S., Woodruff, J. M., & Brennan, M. F. (2000). Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg, 231(1), 51-58.
Demetri, G. D., Reichardt, P., Kang, Y. K., Blay, J. Y., Rutkowski, P., Gelderblom, H., . . . Casali, P. G. (2013). Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet, 381(9863), 295-302. https://doi.org/10.1016/s0140-6736(12)61857-1
Demetri, G. D., Schöffski, P., Grignani, G., Blay, J. Y., Maki, R. G., Van Tine, B. A., . . . Chawla, S. (2017). Activity of Eribulin in Patients With Advanced Liposarcoma Demonstrated in a Subgroup Analysis From a Randomized Phase III Study of Eribulin Versus Dacarbazine. J Clin Oncol, Aug 30 [Epub ahead of print]. https://doi.org/10.1200/jco.2016.71.6605
Demetri, G. D., van Oosterom, A. T., Garrett, C. R., Blackstein, M. E., Shah, M. H., Verweij, J., . . . Casali, P. G. (2006). Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet, 368(9544), 1329-1338.
Demetri, G. D., von Mehren, M., Jones, R. L., Hensley, M. L., Schuetze, S. M., Staddon, A., . . . Patel, S. R. (2016). Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J Clin Oncol, 34(8), 786-793. https://doi.org/10.1200/jco.2015.62.4734
Demizu, Y., Jin, D., Sulaiman, N. S., Nagano, F., Terashima, K., Tokumaru, S., . . . Okimoto, T. (2017). Particle Therapy Using Protons or Carbon Ions for Unresectable or Incompletely Resected Bone and Soft Tissue Sarcomas of the Pelvis. International Journal of Radiation Oncology, Biology, Physics, 98(2), 367-374.
Di Brina, L., Fogliata, A., Navarria, P., D'Agostino, G., Franzese, C., Franceschini, D., . . . Scorsetti, M. (2019). Adjuvant volumetric modulated arc therapy compared to 3D conformal radiation therapy for newly diagnosed soft tissue sarcoma of the extremities: outcome and toxicity evaluation. British Journal of Radiology, 92(1102), 20190252. https://doi.org/10.1259/bjr.20190252
Dickie, C. I., Haas, R., & O'Sullivan, B. (2015). Adjuvant radiation for soft tissue sarcomas. Am Soc Clin Oncol Educ Book, e634-642. https://doi.org/10.14694/EdBook_AM.2015.35.e634
Dirksen, U., Brennan, B., Le Deley, M. C., Cozic, N., van den Berg, H., Bhadri, V., . . . Hawkins, D. S. (2019). High-Dose Chemotherapy Compared With Standard Chemotherapy and Lung Radiation in Ewing Sarcoma With Pulmonary Metastases: Results of the European Ewing Tumour Working Initiative of National Groups, 99 Trial and EWING 2008. J Clin Oncol, 37(34), 3192-3202. https://doi.org/10.1200/jco.19.00915
Duffaud, F., Mir, O., Boudou-Rouquette, P., Piperno-Neumann, S., Penel, N., Bompas, E., . . . Blay, J. Y. (2019). Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: a non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. The Lancet. Oncology, 20(1), 120-133. https://doi.org/10.1016/s1470-2045(18)30742-3
Dyrop, H. B., Vedsted, P., Safwat, A., Maretty-Nielsen, K., Hansen, B. H., Jørgensen, P. H., . . . Keller, J. (2014). Alarm symptoms of soft-tissue and bone sarcoma in patients referred to a specialist center. Acta Orthopaedica, 85(6), 657-662.
Eggermont, A. M., de Wilt, J. H., & ten Hagen, T. L. (2003). Current uses of isolated limb perfusion in the clinic and a model system for new strategies. Lancet Oncology, 4(7), 429-437.
Elvsaas, I.-K. Ø., Graff, B. A., Harboe, I., & Norderhaug, I. N. (2008). Kompresjonsstrømper i forebygging av dyp venetrombose (Rapport fra Kunnskapssenteret nr. 28). Oslo: Nasjonalt kunnskapsenter for helsetjenesten. Hentet fra https://www.fhi.no/publ/eldre/kompresjonsstromper-i-forebygging-av-dyp-venetrombose-/
Emami, B., Lyman, J., Brown, A., Coia, L., Goitein, M., Munzenrider, J. E., . . . Wesson, M. (1991). Tolerance of normal tissue to therapeutic irradiation. Int J Radiat Oncol Biol Phys, 21(1), 109-122.
Emile, J. F., Brahimi, S., Coindre, J. M., Bringuier, P. P., Monges, G., Samb, P., . . . Aegerter, P. (2012). Frequencies of KIT and PDGFRA mutations in the MolecGIST prospective population-based study differ from those of advanced GISTs. Medical Oncology, 29(3), 1765-1772. https://doi.org/10.1007/s12032-011-0074-y
Eng, C., Li, F. P., Abramson, D. H., Ellsworth, R. M., Wong, F. L., Goldman, M. B., . . . Boice, J. D., Jr. (1993). Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst, 85(14), 1121-1128.
Engström, K., Bergh, P., Cederlund, C. G., Hultborn, R., Willen, H., Aman, P., . . . Meis-Kindblom, J. M. (2007). Irradiation of myxoid/round cell liposarcoma induces volume reduction and lipoma-like morphology. Acta oncologica (Stockholm, Sweden), 46(6), 838-845. https://doi.org/10.1080/02841860601080415
Enneking, W. F., Spanier, S. S., & Goodman, M. A. (1980). A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop Relat Res, (153), 106-120.
Eriksson, M. (2010). Histology-driven chemotherapy of soft-tissue sarcoma. Ann Oncol, 21 Suppl 7, vii270-276. https://doi.org/10.1093/annonc/mdq285
Eriksson, M., Reichardt, P., Sundby Hall, K., Schütte, J., Cameron, S., Hohenberger, P., . . . Joensuu, H. (2016). Needle biopsy through the abdominal wall for the diagnosis of gastrointestinal stromal tumour - Does it increase the risk for tumour cell seeding and recurrence? Eur J Cancer, 59, 128-133. https://doi.org/10.1016/j.ejca.2016.02.021
Euramos 1. Randomised trial of the European and American osteosarcoma study group to optimise treatment strategies for resectable osteosarcoma based on histological response to pre-operative chemotherapy. (2012). European and American Osteosarcoma Study Group. Hentet fra http://www.ctu.mrc.ac.uk/euramos/euramos_i_trial.asp
Evrard, R., Schubert, T., Paul, L., & Docquier, P. L. (2019). Resection margins obtained with patient-specific instruments for resecting primary pelvic bone sarcomas: A case-control study. Orthopaedics & Traumatology, Surgery & Research, 105(4), 781-787. https://doi.org/10.1016/j.otsr.2018.12.016
Expert Panel on Musculoskeletal, I., Bestic, J. M., Wessell, D. E., Beaman, F. D., Cassidy, R. C., Czuczman, G. J., . . . Kransdorf, M. J. (2020). ACR Appropriateness Criteria(R) Primary Bone Tumors. J Am Coll Radiol, 17(5S), S226-S238. https://doi.org/10.1016/j.jacr.2020.01.038
FaR-RMS: An Overarching Study for Children and Adults With Frontline and Relapsed RhabdoMyoSarcoma (FaR-RMS). (2020-2030). [pågående studie]. (NCT04625907). Hentet fra https://clinicaltrials.gov/ct2/show/NCT04625907
Fein, D. A., Lee, W. R., Lanciano, R. M., Corn, B. W., Herbert, S. H., Hanlon, A. L., . . . Coia, L. R. (1995). Management of extremity soft tissue sarcomas with limb-sparing surgery and postoperative irradiation: do total dose, overall treatment time, and the surgery-radiotherapy interval impact on local control? Int J Radiat Oncol Biol Phys, 32(4), 969-976.
Feinberg, L., Srinivasan, A., Singh, J. K., Parry, M., Stevenson, J., Jeys, L., . . . Desai, A. (2018). Impact of specialist management on survival from radiation-associated angiosarcoma of the breast. British Journal of Surgery, 105(4), 401-409. https://doi.org/10.1002/bjs.10696
Fendler, W. P., Chalkidis, R. P., Ilhan, H., Knosel, T., Herrmann, K., Issels, R. D., . . . Hacker, M. (2015). Evaluation of several FDG PET parameters for prediction of soft tissue tumour grade at primary diagnosis and recurrence. Eur Radiol, 25(8), 2214-2221. https://doi.org/10.1007/s00330-015-3654-y
Ferrari, A., van Noesel, M. M., Brennan, B., Zanetti, I., Corradini, N., Casanova, M., . . . Orbach, D. (2021). Paediatric non-rhabdomyosarcoma soft tissue sarcomas: the prospective NRSTS 2005 study by the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG). Lancet Child Adolesc Health, 5(8), 546-558. https://doi.org/10.1016/s2352-4642(21)00159-0
Ferrari, S., Bielack, S. S., Smeland, S., Longhi, A., Egerer, G., Sundby Hall, K., . . . Reichardt, P. (2018). EURO-B.O.S.S.: A European study on chemotherapy in bone-sarcoma patients aged over 40: Outcome in primary high-grade osteosarcoma. Tumori, 104(1), 30-36. https://doi.org/10.5301/tj.5000696
Ferrari, S., Smeland, S., Mercuri, M., Bertoni, F., Longhi, A., Ruggieri, P., . . . Scandinavian Sarcoma, G. (2005). Neoadjuvant chemotherapy with high-dose Ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. Journal of Clinical Oncology, 23(34), 8845-8852.
Ferrari, S., Sundby Hall, K., Luksch, R., Tienghi, A., Wiebe, T., Fagioli, F., . . . Smeland, S. (2011). Nonmetastatic Ewing family tumors: high-dose chemotherapy with stem cell rescue in poor responder patients. Results of the Italian Sarcoma Group/Scandinavian Sarcoma Group III protocol. Annals of Oncology, 22(5), 1221-1227.
Ferraro, D., & Zalcberg, J. (2014). Regorafenib in gastrointestinal stromal tumors: clinical evidence and place in therapy. Therapeutic advances in medical oncology, 6(5), 222-228. https://doi.org/10.1177/1758834014544892
Fletcher, C. D., Berman, J. J., Corless, C., Gorstein, F., Lasota, J., Longley, B. J., . . . Weiss, S. W. (2002). Diagnosis of gastrointestinal stromal tumors: a consensus approach. International Journal of Surgical Pathology, 10(2), 81-89.
Forskrift om prioritering av helsetjenester, rett til nødvendig helsehjelp fra spesialisthelsetjenesten, rett til behandling i utlandet og om klagenemnd (prioriteringsforskriften). FOR-2000-12-01-1208. Sist endret i FOR-2021-10-08-2958 fra 01.11.2021. Hentet fra https://lovdata.no/dokument/SF/forskrift/2000-12-01-1208
Frustaci, S., Gherlinzoni, F., De, P. A., Bonetti, M., Azzarelli, A., Comandone, A., . . . Picci, P. (2001). Adjuvant chemotherapy for adult soft tissue sarcomas of the extremities and girdles: results of the Italian randomized cooperative trial. J Clin Oncol, 19(5), 1238-1247.
Fuglø, H. M., Jørgensen, S. M., Loft, A., Hovgaard, D., & Petersen, M. M. (2012). The diagnostic and prognostic value of (1)(8)F-FDG PET/CT in the initial assessment of high-grade bone and soft tissue sarcoma. A retrospective study of 89 patients. European journal of nuclear medicine and molecular imaging, 39(9), 1416-1424. https://doi.org/10.1007/s00259-012-2159-z
Gannon, N. P., King, D. M., Ethun, C. G., Charlson, J., Tran, T. B., Poultsides, G., . . . Bedi, M. (2019). The role of radiation therapy and margin width in localized soft-tissue sarcoma: Analysis from the US Sarcoma Collaborative. Journal of Surgical Oncology, 120(3), 325-331. https://doi.org/10.1002/jso.25522
Garcia del Muro, X. (2015). Nilotinib, imatinib, and GIST therapy. Lancet Oncology, 16(5), 483-484.
Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). (2010). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol, 28(7), 1247-1253. https://doi.org/10.1200/jco.2009.24.2099
Gerbers, J. G., Stevens, M., Ploegmakers, J. J., Bulstra, S. K., & Jutte, P. C. (2014). Computer-assisted surgery in orthopedic oncology. Acta Orthopaedica, 85(6), 663-669.
Gerrand, C., Athanasou, N., Brennan, B., Grimer, R., Judson, I., Morland, B., . . . British Sarcoma, G. (2016). UK guidelines for the management of bone sarcomas. Clinical sarcoma research, 6, 7. https://doi.org/10.1186/s13569-016-0047-1
Gerrand, C. H., Bell, R. S., Wunder, J. S., Kandel, R. A., O'Sullivan, B., Catton, C. N., . . . Davis, A. M. (2003). The influence of anatomic location on outcome in patients with soft tissue sarcoma of the extremity. Cancer, 97(2), 485-492.
Gielen, J. L., De Schepper, A. M., Vanhoenacker, F., Parizel, P. M., Wang, X. L., Sciot, R., & Weyler, J. (2004). Accuracy of MRI in characterization of soft tissue tumors and tumor-like lesions. A prospective study in 548 patients. Eur Radiol, 14(12), 2320-2330.
Giraudet-Le Quintrec, J. S., Coste, J., Vastel, L., Pacault, V., Jeanne, L., Lamas, J. P., . . . Courpied, J. P. (2003). Positive effect of patient education for hip surgery: a randomized trial. Clin Orthop Relat Res, (414), 112-120.
Goldblum, J. R., Weiss, S. W., & Folpe, A. L. (2019). Enzinger and Weiss's soft tissue tumors (7. utg.). Philadelphia: Mosby Elsevier.
Gomarteli, K., Fleckenstein, J., Kirschner, S., Bobu, V., Brockmann, M. A., Henzler, T., . . . Giordano, F. A. (2019). Radiation-induced malignancies after intensity-modulated versus conventional mediastinal radiotherapy in a small animal model. Scientific Reports, 9(1), 15489. https://doi.org/10.1038/s41598-019-51735-3
Griffith, J. F., Yip, S. W. Y., Hung, E. H. Y., Fong, R. C. W., Leung, J., Ng, A. W. H., . . . Lee, R. K. L. (2020). Accuracy of ultrasound in the characterisation of deep soft tissue masses: a prospective study. Eur Radiol, 30(11), 5894-5903. https://doi.org/10.1007/s00330-020-07002-5
Grimer, R. J., Carter, S. R., Tillman, R. M., & Abudu, A. (2001). Early symptoms and diagnosis of bone tumors. J Bone Joint Surg Am, 83-A(7), 1107-1108.
Grimer, R. J., & Sneath, R. S. (1990). Diagnosing malignant bone tumours. J Bone Joint Surg Br, 72(5), 754-756.
Gronchi, A., Bonvalot, S., Poveda Velasco, A., Kotasek, D., Rutkowski, P., Hohenberger, P., . . . Casali, P. G. (2020). Quality of Surgery and Outcome in Localized Gastrointestinal Stromal Tumors Treated Within an International Intergroup Randomized Clinical Trial of Adjuvant Imatinib. JAMA Surg, 155(6), e200397. https://doi.org/10.1001/jamasurg.2020.0397
Gronchi, A., Lo Vullo, S., Fiore, M., Mussi, C., Stacchiotti, S., Collini, P., . . . Casali, P. G. (2009). Aggressive surgical policies in a retrospectively reviewed single-institution case series of retroperitoneal soft tissue sarcoma patients. J Clin Oncol, 27(1), 24-30. https://doi.org/10.1200/jco.2008.17.8871
Gronchi, A., Miah, A. B., Dei Tos, A. P., Abecassis, N., Bajpai, J., Bauer, S., . . . Stacchiotti, S. (2021). Soft tissue and visceral sarcomas: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up(☆). Ann Oncol, 32(11), 1348-1365. https://doi.org/10.1016/j.annonc.2021.07.006
Gronchi, A., Miceli, R., Allard, M. A., Callegaro, D., Le Péchoux, C., Fiore, M., . . . Bonvalot, S. (2015). Personalizing the approach to retroperitoneal soft tissue sarcoma: histology-specific patterns of failure and postrelapse outcome after primary extended resection. Ann Surg Oncol, 22(5), 1447-1454. https://doi.org/10.1245/s10434-014-4130-7
Gronchi, A., Palmerini, E., Quagliuolo, V., Martin Broto, J., Lopez Pousa, A., Grignani, G., . . . Casali, P. G. (2020). Neoadjuvant Chemotherapy in High-Risk Soft Tissue Sarcomas: Final Results of a Randomized Trial From Italian (ISG), Spanish (GEIS), French (FSG), and Polish (PSG) Sarcoma Groups. J Clin Oncol, 38(19), 2178-2186. https://doi.org/10.1200/jco.19.03289
Gronchi, A., Strauss, D. C., Miceli, R., Bonvalot, S., Swallow, C. J., Hohenberger, P., . . . Raut, C. P. (2016). Variability in Patterns of Recurrence After Resection of Primary Retroperitoneal Sarcoma (RPS): A Report on 1007 Patients From the Multi-institutional Collaborative RPS Working Group. Ann Surg, 263(5), 1002-1009. https://doi.org/10.1097/sla.0000000000001447
Grosso, F., Sanfilippo, R., Virdis, E., Piovesan, C., Collini, P., Dileo, P., . . . Casali, P. G. (2009). Trabectedin in myxoid liposarcomas (MLS): a long-term analysis of a single-institution series. Ann Oncol, 20(8), 1439-1444.
Gustafson, P., Dreinhofer, K. E., & Rydholm, A. (1994). Soft tissue sarcoma should be treated at a tumor center. A comparison of quality of surgery in 375 patients. Acta Orthop Scand, 65(1), 47-50.
Gustafsson, U. O., Scott, M. J., Schwenk, W., Demartines, N., Roulin, D., Francis, N., . . . Nutrition. (2013). Guidelines for perioperative care in elective colonic surgery: Enhanced Recovery After Surgery (ERAS()) Society recommendations. World Journal of Surgery, 37(2), 259-284.
Gyorki, D. E., & Brennan, M. F. (2014). Management of recurrent retroperitoneal sarcoma. Journal of Surgical Oncology, 109(1), 53-59. https://doi.org/10.1002/jso.23463
Habrand, J. L., Schneider, R., Alapetite, C., Feuvret, L., Petras, S., Datchary, J., . . . Sainte-Rose, C. (2008). Proton therapy in pediatric skull base and cervical canal low-grade bone malignancies. Int J Radiat Oncol Biol Phys, 71(3), 672-675. https://doi.org/10.1016/j.ijrobp.2008.02.043
Haibach, H., Farrell, C., & Dittrich, F. J. (1985). Neoplasms arising in Paget's disease of bone: a study of 82 cases. Am J Clin Pathol, 83(5), 594-600.
Hall, K. S., Bruland Ø, S., Bjerkehagen, B., Lidbrink, E., Jebsen, N., Hagberg, H., . . . Eriksson, M. (2020). Preoperative accelerated radiotherapy combined with chemotherapy in a defined cohort of patients with high risk soft tissue sarcoma: a Scandinavian Sarcoma Group study. Clinical sarcoma research, 10(1), 22. https://doi.org/10.1186/s13569-020-00145-5
Hardes, J., von Eiff, C., Streitbuerger, A., Balke, M., Budny, T., Henrichs, M. P., . . . Ahrens, H. (2010). Reduction of periprosthetic infection with silver-coated megaprostheses in patients with bone sarcoma. Journal of Surgical Oncology, 101(5), 389-395. https://doi.org/10.1002/jso.21498
Hartmann, J. T., Kopp, H. G., Gruenwald, V., Piperno-Neumann, S., Kunitz, A., Hofheinz, R., . . . Mayer, F. (2020). Randomised phase II trial of trofosfamide vs. doxorubicin in elderly patients with untreated metastatic soft-tissue sarcoma. Eur J Cancer, 124, 152-160. https://doi.org/10.1016/j.ejca.2019.10.016
Hawkins, M. M., Wilson, L. M., Burton, H. S., Potok, M. H., Winter, D. L., Marsden, H. B., & Stovall, M. A. (1996). Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J Natl Cancer Inst, 88(5), 270-278.
Helse- og omsorgsdepartementet. (2015). Nasjonal helse- og sykehusplan (2016–2019) (Meld. St. 11 (2015–2016)). Hentet fra https://www.regjeringen.no/no/dokumenter/meld.-st.-11-20152016/id2462047/
Hensley, M. L., Maki, R., Venkatraman, E., Geller, G., Lovegren, M., Aghajanian, C., . . . Spriggs, D. R. (2002). Gemcitabine and docetaxel in patients with unresectable leiomyosarcoma: results of a phase II trial. J Clin Oncol, 20(12), 2824-2831.
Heslin, M. J., Lewis, J. J., Nadler, E., Newman, E., Woodruff, J. M., Casper, E. S., . . . Brennan, M. F. (1997). Prognostic factors associated with long-term survival for retroperitoneal sarcoma: implications for management. J Clin Oncol, 15(8), 2832-2839.
Higuchi, T., Yamamoto, N., Nishida, H., Hayashi, K., Takeuchi, A., Kimura, H., . . . Tsuchiya, H. (2017). Knee joint preservation surgery in osteosarcoma using tumour-bearing bone treated with liquid nitrogen. International orthopaedics, Jun 1 [Epub ahead of print]. https://doi.org/10.1007/s00264-017-3499-x
Hompland, I., Ferrari, S., Bielack, S., Palmerini, E., Hall, K. S., Picci, P., . . . Smeland, S. (2021). Outcome in dedifferentiated chondrosarcoma for patients treated with multimodal therapy: Results from the EUROpean Bone Over 40 Sarcoma Study. Eur J Cancer, 151, 150-158. https://doi.org/10.1016/j.ejca.2021.04.017
Hongtao, L., Hui, Z., Bingshun, W., Xiaojin, W., Zhiyu, W., Shuier, Z., . . . Yang, Y. (2012). 18F-FDG positron emission tomography for the assessment of histological response to neoadjuvant chemotherapy in osteosarcomas: a meta-analysis. Surgical Oncology, 21(4), e165-170. https://doi.org/10.1016/j.suronc.2012.07.002
Hough, A. (2001). Physiotherapy in respiratory care: an evidence-based approach to respiratory and cardiac management. Cheltenham: Trans-Atlantic Publications.
Huang, T., Li, F., Yan, Z., Ma, Y., Xiong, F., Cai, X., . . . Dong, J. (2018). Effectiveness of 18F-FDG PET/CT in the diagnosis, staging and recurrence monitoring of Ewing sarcoma family of tumors: A meta-analysis of 23 studies. Medicine (Baltimore), 97(48), e13457. https://doi.org/10.1097/MD.0000000000013457
Hug, E. B., Adams, J., Fitzek, M., De Vries, A., & Munzenrider, J. E. (2000). Fractionated, three-dimensional, planning-assisted proton-radiation therapy for orbital rhabdomyosarcoma: a novel technique. Int J Radiat Oncol Biol Phys, 47(4), 979-984.
Hung, E. H. Y., Griffith, J. F., Yip, S. W. Y., Ivory, M., Lee, J. C. H., Ng, A. W. H., & Tong, C. S. L. (2020). Accuracy of ultrasound in the characterization of superficial soft tissue tumors: a prospective study. Skeletal Radiology, 49(6), 883-892. https://doi.org/10.1007/s00256-019-03365-z
Hung, J., & Anderson, R. (1997). p53: functions, mutations and sarcomas. Acta Orthop Scand suppl, 273, 68-73.
Huvos, A. G., Butler, A., & Bretsky, S. S. (1983). Osteogenic sarcoma associated with Paget's disease of bone. A clinicopathologic study of 65 patients. Cancer, 52(8), 1489-1495.
Huvos, A. G., Sundaresan, N., Bretsky, S. S., & Butler, A. (1985). Osteogenic sarcoma of the skull. A clinicopathologic study of 19 patients. Cancer, 56(5), 1214-1221.
Hwang, J. P., Lim, I., Kong, C. B., Jeon, D. G., Byun, B. H., Kim, B. I., . . . Lim, S. M. (2016). Prognostic Value of SUVmax Measured by Pretreatment Fluorine-18 Fluorodeoxyglucose Positron Emission Tomography/Computed Tomography in Patients with Ewing Sarcoma. PloS One, 11(4), e0153281. https://doi.org/10.1371/journal.pone.0153281
Hølmebakk, T., Bjerkehagen, B., Hompland, I., Stoldt, S., & Boye, K. (2019). Relationship between R1 resection, tumour rupture and recurrence in resected gastrointestinal stromal tumour. British Journal of Surgery, 106(4), 419-426. https://doi.org/10.1002/bjs.11027
Hølmebakk, T., Hompland, I., Bjerkehagen, B., Stoldt, S., Bruland Ø, S., Hall, K. S., & Boye, K. (2018). Recurrence-Free Survival After Resection of Gastric Gastrointestinal Stromal Tumors Classified According to a Strict Definition of Tumor Rupture: A Population-Based Study. Ann Surg Oncol, 25(5), 1133-1139. https://doi.org/10.1245/s10434-018-6353-5
Hølmebakk, T., Wiedswang, A. M., Meza-Zepeda, L. A., Hompland, I., Lobmaier, I. V. K., Berner, J. M., . . . Boye, K. (2021). Integrating Anatomical, Molecular and Clinical Risk Factors in Gastrointestinal Stromal Tumor of the Stomach. Ann Surg Oncol, 28(11), 6837-6845. https://doi.org/10.1245/s10434-021-09605-8
Haas, R. L., Delaney, T. F., O'Sullivan, B., Keus, R. B., Le Pechoux, C., Olmi, P., . . . Wang, D. (2012). Radiotherapy for management of extremity soft tissue sarcomas: why, when, and where? Int J Radiat Oncol Biol Phys, 84(3), 572-580. https://doi.org/10.1016/j.ijrobp.2012.01.062
Igaki, H., Tokuuye, K., Okumura, T., Sugahara, S., Kagei, K., Hata, M., . . . Akine, Y. (2004). Clinical results of proton beam therapy for skull base chordoma. Int J Radiat Oncol Biol Phys, 60(4), 1120-1126. https://doi.org/10.1016/j.ijrobp.2004.05.064
Igarashi, K., Yamamoto, N., Shirai, T., Hayashi, K., Nishida, H., Kimura, H., . . . Tsuchiya, H. (2014). The long-term outcome following the use of frozen autograft treated with liquid nitrogen in the management of bone and soft-tissue sarcomas. The bone & joint journal, 96-B(4), 555-561. https://doi.org/10.1302/0301-620X.96B4.32629
Issels, R., & Lindner, L. H. (2016). Regional hyperthermia for high-risk soft tissue sarcoma treatment: present status and next questions. Current Opinion in Oncology, 28(5), 447-452.
Issels, R. D., Lindner, L. H., Verweij, J., Wust, P., Reichardt, P., Schem, B. C., . . . European Society for Hyperthermic, O. (2010). Neo-adjuvant chemotherapy alone or with regional hyperthermia for localised high-risk soft-tissue sarcoma: a randomised phase 3 multicentre study. Lancet Oncology, 11(6), 561-570.
An italian - scandinavian treatment protocol for metastatic and pelvic osteosarcoma (ISG/SSG II). (2002). Bologna; Lund: Italian sarcoma group (ISG), Scandinavian sarcoma group and oncologic center (SSG).
Jebsen, N. L., Bruland, Ø. S., Eriksson, M., Engellau, J., Turesson, I., Folin, A., . . . Hall, K. S. (2011). Five-year results from a Scandinavian sarcoma group study (SSG XIII) of adjuvant chemotherapy combined with accelerated radiotherapy in high-risk soft tissue sarcoma of extremities and trunk wall. Int J Radiat Oncol Biol Phys, 81(5), 1359-1366. https://doi.org/10.1016/j.ijrobp.2010.07.037
Jebsen, N. L., Trovik, C. S., Bauer, H. C., Rydholm, A., Monge, O. R., Hall, K. S., . . . Bruland, O. S. (2008). Radiotherapy to improve local control regardless of surgical margin and malignancy grade in extremity and trunk wall soft tissue sarcoma: a Scandinavian sarcoma group study. Int J Radiat Oncol Biol Phys, 71(4), 1196-1203.
Jian, B. J., Bloch, O. G., Yang, I., Han, S. J., Aranda, D., Tihan, T., & Parsa, A. T. (2010). Adjuvant radiation therapy and chondroid chordoma subtype are associated with a lower tumor recurrence rate of cranial chordoma. J Neurooncol, 98(1), 101-108.
Jingu, K., Tsujii, H., Mizoe, J. E., Hasegawa, A., Bessho, H., Takagi, R., . . . Organizing Committee for the Working Group for Head-and-Neck, C. (2012). Carbon ion radiation therapy improves the prognosis of unresectable adult bone and soft-tissue sarcoma of the head and neck. International Journal of Radiation Oncology, Biology, Physics, 82(5), 2125-2131.
Joensuu, H. (2008). Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol, 39(10), 1411-1419.
Joensuu, H., Eriksson, M., Collan, J., Balk, M. H., Leyvraz, S., & Montemurro, M. (2015). Radiotherapy for GIST progressing during or after tyrosine kinase inhibitor therapy: A prospective study. Radiother Oncol, 116(2), 233-238. https://doi.org/10.1016/j.radonc.2015.07.025
Joensuu, H., Eriksson, M., Sundby Hall, K., Hartmann, J. T., Pink, D., Schutte, J., . . . Reichardt, P. (2012). One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA, 307(12), 1265-1272. https://doi.org/10.1001/jama.2012.347
Joensuu, H., Eriksson, M., Sundby Hall, K., Reichardt, A., Hartmann, J. T., Pink, D., . . . Reichardt, P. (2016). Adjuvant Imatinib for High-Risk GI Stromal Tumor: Analysis of a Randomized Trial. Journal of Clinical Oncology, 34(3), 244-250.
Joensuu, H., Eriksson, M., Sundby Hall, K., Reichardt, A., Hermes, B., Schütte, J., . . . Reichardt, P. (2020). Survival Outcomes Associated With 3 Years vs 1 Year of Adjuvant Imatinib for Patients With High-Risk Gastrointestinal Stromal Tumors: An Analysis of a Randomized Clinical Trial After 10-Year Follow-up. JAMA Oncol, 6(8), 1241-1246. https://doi.org/10.1001/jamaoncol.2020.2091
Joensuu, H., Hohenberger, P., & Corless, C. L. (2013). Gastrointestinal stromal tumour. Lancet, 382(9896), 973-983. https://doi.org/10.1016/s0140-6736(13)60106-3
Joensuu, H., Vehtari, A., Riihimaki, J., Nishida, T., Steigen, S. E., Brabec, P., . . . Rutkowski, P. (2012). Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. The Lancet. Oncology, 13(3), 265-274. https://doi.org/10.1016/s1470-2045(11)70299-6
Joensuu, H., Wardelmann, E., Sihto, H., Eriksson, M., Sundby Hall, K., Reichardt, A., . . . Reichardt, P. (2017). Effect of KIT and PDGFRA Mutations on Survival in Patients With Gastrointestinal Stromal Tumors Treated With Adjuvant Imatinib: An Exploratory Analysis of a Randomized Clinical Trial. JAMA Oncol, 3(5), 602-609. https://doi.org/10.1001/jamaoncol.2016.5751
Jones, R. L., Serrano, C., von Mehren, M., George, S., Heinrich, M. C., Kang, Y. K., . . . Bauer, S. (2021). Avapritinib in unresectable or metastatic PDGFRA D842V-mutant gastrointestinal stromal tumours: Long-term efficacy and safety data from the NAVIGATOR phase I trial. Eur J Cancer, 145, 132-142. https://doi.org/10.1016/j.ejca.2020.12.008
Jordhøy, M. S., Fayers, P., Loge, J. H., Ahlner-Elmqvist, M., & Kaasa, S. (2001). Quality of life in palliative cancer care: results from a cluster randomized trial. J Clin Oncol, 19(18), 3884-3894.
Judson, I., Verweij, J., Gelderblom, H., Hartmann, J. T., Schöffski, P., Blay, J. Y., . . . van der Graaf, W. T. (2014). Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. The Lancet. Oncology, 15(4), 415-423. https://doi.org/10.1016/s1470-2045(14)70063-4
Kamada, T., Tsujii, H., Tsuji, H., Yanagi, T., Mizoe, J. E., Miyamoto, T., . . . Soft Tissue, S. (2002). Efficacy and safety of carbon ion radiotherapy in bone and soft tissue sarcomas. Journal of Clinical Oncology, 20(22), 4466-4471.
Kang, Y. K., Ryu, M. H., Yoo, C., Ryoo, B. Y., Kim, H. J., Lee, J. J., . . . Demetri, G. D. (2013). Resumption of imatinib to control metastatic or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib (RIGHT): a randomised, placebo-controlled, phase 3 trial. The Lancet. Oncology, 14(12), 1175-1182. https://doi.org/10.1016/s1470-2045(13)70453-4
Kepka, L., DeLaney, T. F., Suit, H. D., & Goldberg, S. I. (2005). Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys, 63(3), 852-859.
Kinge, B., Tranheim, R. S., & Eide, N. A. (2004). Retinoblastom--arvelig øyekreft hos barn. [Retinoblastoma--hereditary eye cancer in children .]. Tidsskr Nor Lægeforen, 124(2), 183-185.
Koontz, B. F., Clough, R. W., & Halperin, E. C. (2006). Palliative radiation therapy for metastatic Ewing sarcoma. Cancer, 106(8), 1790-1793.
Kransdorf, M. J. (2009). Society of skeletal radiology 2008 annual meeting. Skeletal Radiology, 38(1), 97-100.
Kransdorf, M. J., & Murphey, M. D. (2000). Radiologic evaluation of soft-tissue masses: a current perspective. AJR Am J Roentgenol, 175(3), 575-587.
Kransdorf, M. J., & Murphey, M. D. (2006). Imaging of soft tissue tumors (2. utg.). Philadelphia, Pa.: Lippincott Williams & Wilkins.
Kubo, T., Furuta, T., Johan, M. P., & Ochi, M. (2016). Prognostic significance of (18)F-FDG PET at diagnosis in patients with soft tissue sarcoma and bone sarcoma; systematic review and meta-analysis. European Journal of Cancer, 58, 104-111.
Ladenstein, R., Pötschger, U., Le Deley, M. C., Whelan, J., Paulussen, M., Oberlin, O., . . . Jürgens, H. (2010). Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol, 28(20), 3284-3291. https://doi.org/10.1200/jco.2009.22.9864
Lagrange, J. L., Ramaioli, A., Chateau, M. C., Marchal, C., Resbeut, M., Richaud, P., . . . Coindre, J. M. (2000). Sarcoma after radiation therapy: retrospective multiinstitutional study of 80 histologically confirmed cases. Radiation Therapist and Pathologist Groups of the Federation Nationale des Centres de Lutte Contre le Cancer. Radiology, 216(1), 197-205.
Lakkaraju, A., Sinha, R., Garikipati, R., Edward, S., & Robinson, P. (2009). Ultrasound for initial evaluation and triage of clinically suspicious soft-tissue masses. Clinical Radiology, 64(6), 615-621. https://doi.org/10.1016/j.crad.2009.01.012
Lansu, J., Bovée, J., Braam, P., van Boven, H., Flucke, U., Bonenkamp, J. J., . . . Haas, R. L. (2021). Dose Reduction of Preoperative Radiotherapy in Myxoid Liposarcoma: A Nonrandomized Controlled Trial. JAMA Oncol, 7(1), e205865. https://doi.org/10.1001/jamaoncol.2020.5865
Le Cesne, A., Antoine, E., Spielmann, M., Le Chevalier, T., Brain, E., Toussaint, C., . . . et al. (1995). High-dose ifosfamide: circumvention of resistance to standard-dose ifosfamide in advanced soft tissue sarcomas. J Clin Oncol, 13(7), 1600-1608. https://doi.org/10.1200/jco.1995.13.7.1600
Le Cesne, A., Blay, J. Y., Judson, I., Van, O. A., Verweij, J., Radford, J., . . . Nielsen, O. S. (2005). Phase II study of ET-743 in advanced soft tissue sarcomas: a European Organisation for the Research and Treatment of Cancer (EORTC) soft tissue and bone sarcoma group trial. J Clin Oncol, 23(3), 576-584.
Le Cesne, A., Ray-Coquard, I., Bui, B. N., Adenis, A., Rios, M., Bertucci, F., . . . Blay, J. Y. (2010). Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomised phase 3 trial. The Lancet. Oncology, 11(10), 942-949. https://doi.org/10.1016/s1470-2045(10)70222-9
Lee, A. Y., Rickles, F. R., Julian, J. A., Gent, M., Baker, R. I., Bowden, C., . . . Levine, M. N. (2005). Randomized comparison of low molecular weight heparin and coumarin derivatives on the survival of patients with cancer and venous thromboembolism. J Clin Oncol, 23(10), 2123-2129.
Lee, C. T., Bilton, S. D., Famiglietti, R. M., Riley, B. A., Mahajan, A., Chang, E. L., . . . Smith, A. R. (2005). Treatment planning with protons for pediatric retinoblastoma, medulloblastoma, and pelvic sarcoma: how do protons compare with other conformal techniques? Int J Radiat Oncol Biol Phys, 63(2), 362-372. https://doi.org/10.1016/j.ijrobp.2005.01.060
Levernes, S., & Johannessen, D. C. (2003). Volum og doser ved strålebehandling: definisjoner, retningslinjer for bruk, dokumentasjon og rapportering (StrålevernRapport 2003:12). Østerås: Statens Strålevern. Hentet fra https://dsa.no/publikasjoner/stralevernrapport-12-2003-volum-og-doser-ved-stralebehandling/straalevernrapport-2003-12-volum-og-doser-ved-straalebehandling-definisjoner-retningslinjer-dokumentasjon-rapportering.pdf
Lewis, J. J., Leung, D., Woodruff, J. M., & Brennan, M. F. (1998). Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg, 228(3), 355-365.
Li, F. P., & Fraumeni, J. F., Jr. (1969). Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med, 71(4), 747-752.
Li, Y. J., Dai, Y. L., Cheng, Y. S., Zhang, W. B., & Tu, C. Q. (2016). Positron emission tomography (18)F-fluorodeoxyglucose uptake and prognosis in patients with bone and soft tissue sarcoma: A meta-analysis. Eur J Surg Oncol, 42(8), 1103-1114. https://doi.org/10.1016/j.ejso.2016.04.056
Lim, H. J., Johnny Ong, C. A., Tan, J. W., & Ching Teo, M. C. (2019). Utility of positron emission tomography/computed tomography (PET/CT) imaging in the evaluation of sarcomas: A systematic review. Critical Reviews in Oncology/Hematology, 143, 1-13. https://doi.org/10.1016/j.critrevonc.2019.07.002
Liu, F., Zhang, Q., Zhou, D., & Dong, J. (2019). Effectiveness of (18)F-FDG PET/CT in the diagnosis and staging of osteosarcoma: a meta-analysis of 26 studies. BMC Cancer, 19(1), 323. https://doi.org/10.1186/s12885-019-5488-5
Liu, J., Hudkins, P. G., Swee, R. G., & Unni, K. K. (1987). Bone sarcomas associated with Ollier's disease. Cancer, 59(7), 1376-1385.
Longhi, A., Paioli, A., Palmerini, E., Cesari, M., Abate, M. E., Setola, E., . . . Boye, K. (2019). Pazopanib in relapsed osteosarcoma patients: report on 15 cases. Acta oncologica (Stockholm, Sweden), 58(1), 124-128. https://doi.org/10.1080/0284186x.2018.1503714
Lov om folketrygd (folketrygdloven). LOV-1997-02-28-19. Sist endret i LOV-2021-12-22-151 fra 01.01.2022. Hentet fra https://lovdata.no/dokument/NL/lov/1997-02-28-19
Lov om pasient- og brukerrettigheter (pasient- og brukerrettighetsloven). LOV-1999-07-02-63. Sist endret i LOV-2021-05-07-31 fra 01.07.2021. Hentet fra https://lovdata.no/dokument/NL/lov/1999-07-02-63
Luksch, R., Tienghi, A., Hall, K. S., Fagioli, F., Picci, P., Barbieri, E., . . . Ferrari, S. (2012). Primary metastatic Ewing's family tumors: results of the Italian Sarcoma Group and Scandinavian Sarcoma Group ISG/SSG IV Study including myeloablative chemotherapy and total-lung irradiation. Ann Oncol, 23(11), 2970-2976. https://doi.org/10.1093/annonc/mds117
Ma, L. D., McCarthy, E. F., Bluemke, D. A., & Frassica, F. J. (1998). Differentiation of benign from malignant musculoskeletal lesions using MR imaging: pitfalls in MR evaluation of lesions with a cystic appearance. AJR Am J Roentgenol, 170(5), 1251-1258.
Macpherson, R. E., Pratap, S., Tyrrell, H., Khonsari, M., Wilson, S., Gibbons, M., . . . Hassan, A. B. (2018). Retrospective audit of 957 consecutive (18)F-FDG PET-CT scans compared to CT and MRI in 493 patients with different histological subtypes of bone and soft tissue sarcoma. Clinical sarcoma research, 8, 9. https://doi.org/10.1186/s13569-018-0095-9
Malkin, D., Jolly, K. W., Barbier, N., Look, A. T., Friend, S. H., Gebhardt, M. C., . . . . (1992). Germline mutations of the p53 tumor-suppressor gene in children and young adults with second malignant neoplasms. N Engl J Med, 326(20), 1309-1315.
Mankin, H. J., Lange, T. A., & Spanier, S. S. (1982). The hazards of biopsy in patients with malignant primary bone and soft-tissue tumors. J Bone Joint Surg Am, 64(8), 1121-1127.
Marina, N. M., Smeland, S., Bielack, S. S., Bernstein, M., Jovic, G., Krailo, M. D., . . . Whelan, J. S. (2016). Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): an open-label, international, randomised controlled trial. Lancet Oncology, 17(10), 1396-1408.
Markhede, G., & Stener, B. (1981). Function after removal of various hip and thigh muscles for extirpation of tumors. Acta Orthop Scand, 52(4), 373-395.
Marks, L. B., Yorke, E. D., Jackson, A., Ten Haken, R. K., Constine, L. S., Eisbruch, A., . . . Deasy, J. O. (2010). Use of normal tissue complication probability models in the clinic. Int J Radiat Oncol Biol Phys, 76(3 Suppl), S10-19. https://doi.org/10.1016/j.ijrobp.2009.07.1754
Menu-Branthomme, A., Rubino, C., Shamsaldin, A., Hawkins, M. M., Grimaud, E., Dondon, M. G., . . . de Vathaire, F. (2004). Radiation dose, chemotherapy and risk of soft tissue sarcoma after solid tumours during childhood. International Journal of Cancer, 110(1), 87-93.
Meyers, P. A., Schwartz, C. L., Krailo, M. D., Healey, J. H., Bernstein, M. L., Betcher, D., . . . Children's Oncology, G. (2008). Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival--a report from the Children's Oncology Group. Journal of Clinical Oncology, 26(4), 633-638.
Miettinen, M., & Lasota, J. (2006). Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol, 23(2), 70-83. https://doi.org/10.1053/j.semdp.2006.09.001
Milano, M. T., Constine, L. S., & Okunieff, P. (2007). Normal tissue tolerance dose metrics for radiation therapy of major organs. Semin Radiat Oncol, 17(2), 131-140.
Miller, T. T. (2008). Bone tumors and tumorlike conditions: analysis with conventional radiography. Radiology, 246(3), 662-674.
Mir, O., Cropet, C., Toulmonde, M., Cesne, A. L., Molimard, M., Bompas, E., . . . Osseuses, P. s. g. o. t. F. S. G.-G. d. E. d. T. (2016). Pazopanib plus best supportive care versus best supportive care alone in advanced gastrointestinal stromal tumours resistant to imatinib and sunitinib (PAZOGIST): a randomised, multicentre, open-label phase 2 trial. The Lancet. Oncology, 17(5), 632-641. https://doi.org/10.1016/S1470-2045(16)00075-9
Miralbell, R., Lomax, A., Cella, L., & Schneider, U. (2002). Potential reduction of the incidence of radiation-induced second cancers by using proton beams in the treatment of pediatric tumors. Int J Radiat Oncol Biol Phys, 54(3), 824-829.
Mizumoto, M., Murayama, S., Akimoto, T., Demizu, Y., Fukushima, T., Ishida, Y., . . . Sakurai, H. (2018). Preliminary results of proton radiotherapy for pediatric rhabdomyosarcoma: a multi-institutional study in Japan. Cancer Med, 7(5), 1870-1874. https://doi.org/10.1002/cam4.1464
Morgan, E. A., Kozono, D. E., Wang, Q., Mery, C. M., Butrynski, J. E., Baldini, E. H., . . . Raut, C. P. (2012). Cutaneous radiation-associated angiosarcoma of the breast: poor prognosis in a rare secondary malignancy. Ann Surg Oncol, 19(12), 3801-3808. https://doi.org/10.1245/s10434-012-2563-4
Moser, R. P., Jr., & Madewell, J. E. (1987). An approach to primary bone tumors. Radiol Clin North Am, 25(6), 1049-1093.
Moulton, J. S., Blebea, J. S., Dunco, D. M., Braley, S. E., Bisset, G. S., III, & Emery, K. H. (1995). MR imaging of soft-tissue masses: diagnostic efficacy and value of distinguishing between benign and malignant lesions. AJR Am J Roentgenol, 164(5), 1191-1199.
Mucciarini, C., Rossi, G., Bertolini, F., Valli, R., Cirilli, C., Rashid, I., . . . Federico, M. (2007). Incidence and clinicopathologic features of gastrointestinal stromal tumors. A population-based study. BMC Cancer, 7, 230. https://doi.org/10.1186/1471-2407-7-230
Murray, E. M., Werner, D., Greeff, E. A., & Taylor, D. A. (1999). Postradiation sarcomas: 20 cases and a literature review. Int J Radiat Oncol Biol Phys, 45(4), 951-961.
Nasjonale faglige retningslinjer for forebygging og behandling av underernæring. (2009). (IS-1580). Oslo: Helsedirektoratet. Hentet fra http://helsedirektoratet.no/publikasjoner/nasjonal-faglig-retningslinje-for-forebygging-og-behandling-av-underernering/Publikasjoner/nasjonal-faglig-retningslinje-for-forebygging-og-behandling-av-underernering.PDF
Nasjonalt handlingsprogram for palliasjon i kreftomsorgen: nasjonal faglig retningslinje. (2019). (rev utg.). (IS-2800). Oslo: Helsedirektoratet. Hentet fra https://www.helsedirektoratet.no/retningslinjer/palliasjon-i-kreftomsorgen-handlingsprogram
Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft hos barn: nasjonal faglig retningslinje. (2020). (IS-2925). Oslo: Helsedirektoratet. Hentet fra https://www.helsedirektoratet.no/retningslinjer/kreft-hos-barn-handlingsprogram
Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne lymfomer. (2012). (rev utg.). (IS-1594). Oslo: Helsedirektoratet. Hentet fra http://www.helsedirektoratet.no/publikasjoner/nasjonalt-handlingsprogram-med-retningslinjer-for-diagnostikk-behandling-og-oppfolging-av-maligne-lymfomer/Sider/default.aspx
Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av pasienter med brystkreft: nasjonal faglig retningslinje. (2021). (rev. utg.). (IS-3012). Oslo: Helsedirektoratet. Hentet fra https://www.helsedirektoratet.no/retningslinjer/brystkreft-handlingsprogram
Noel, G., Feuvret, L., Calugaru, V., Dhermain, F., Mammar, H., Haie-Meder, C., . . . Mazeron, J. J. (2005). Chordomas of the base of the skull and upper cervical spine. One hundred patients irradiated by a 3D conformal technique combining photon and proton beams. Acta oncologica (Stockholm, Sweden), 44(7), 700-708. https://doi.org/10.1080/02841860500326257
Noel, G., Habrand, J. L., Jauffret, E., de Crevoisier, R., Dederke, S., Mammar, H., . . . Mazeron, J. J. (2003). Radiation therapy for chordoma and chondrosarcoma of the skull base and the cervical spine. Prognostic factors and patterns of failure. Strahlentherapie und Onkologie : Organ der Deutschen Rontgengesellschaft ... [et al], 179(4), 241-248. https://doi.org/10.1007/s00066-003-1065-5
Nordal, R. R. (1998). Uterine sarcomas in Norway 1956-1992: an epidemiological and clinicopathological study (doktorgrad). Oslo: Faculty of Medicine, University of Oslo.
O'Sullivan, B., Davis, A. M., Turcotte, R., Bell, R., Catton, C., Chabot, P., . . . Zee, B. (2002). Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: a randomised trial. Lancet, 359(9325), 2235-2241.
O'Sullivan, B., Wylie, J., Catton, C., Gutierrez, E., Swallow, C. J., Wunder, J., . . . Bell, R. (1999). The local management of soft tissue sarcoma. Semin Radiat Oncol, 9(4), 328-348.
Oberlin, O., Rey, A., Desfachelles, A. S., Philip, T., Plantaz, D., Schmitt, C., . . . Michon, J. (2006). Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors: a study by the Société Française des Cancers de l'Enfant. J Clin Oncol, 24(24), 3997-4002. https://doi.org/10.1200/jco.2006.05.7059
Palmerini, E., Colangeli, M., Nanni, C., Fanti, S., Marchesi, E., Paioli, A., . . . Ferrari, S. (2017). The role of FDG PET/CT in patients treated with neoadjuvant chemotherapy for localized bone sarcomas. European journal of nuclear medicine and molecular imaging, 44(2), 215-223. https://doi.org/10.1007/s00259-016-3509-z
Palmerini, E., Jones, R. L., Marchesi, E., Paioli, A., Cesari, M., Longhi, A., . . . Ferrari, S. (2016). Gemcitabine and docetaxel in relapsed and unresectable high-grade osteosarcoma and spindle cell sarcoma of bone. BMC Cancer, 16, 280. https://doi.org/10.1186/s12885-016-2312-3
Parker, W. H., Fu, Y. S., & Berek, J. S. (1994). Uterine sarcoma in patients operated on for presumed leiomyoma and rapidly growing leiomyoma. Obstet Gynecol, 83(3), 414-418.
Pasquali, S., Pizzamiglio, S., Touati, N., Litiere, S., Marreaud, S., Kasper, B., . . . Gronchi, A. (2019). The impact of chemotherapy on survival of patients with extremity and trunk wall soft tissue sarcoma: revisiting the results of the EORTC-STBSG 62931 randomised trial. Eur J Cancer, 109, 51-60. https://doi.org/10.1016/j.ejca.2018.12.009
Penel, N., Bui, B. N., Bay, J. O., Cupissol, D., Ray-Coquard, I., Piperno-Neumann, S., . . . Blay, J. Y. (2008). Phase II trial of weekly paclitaxel for unresectable angiosarcoma: the ANGIOTAX Study. J Clin Oncol, 26(32), 5269-5274.
Pervaiz, N., Colterjohn, N., Farrokhyar, F., Tozer, R., Figueredo, A., & Ghert, M. (2008). A systematic meta-analysis of randomized controlled trials of adjuvant chemotherapy for localized resectable soft-tissue sarcoma. Cancer, 113(3), 573-581.
Petasnick, J. P., Turner, D. A., Charters, J. R., Gitelis, S., & Zacharias, C. E. (1986). Soft-tissue masses of the locomotor system: comparison of MR imaging with CT. Radiology, 160(1), 125-133.
Physiotherapy for respiratory and cardiac problems: adults and paediatrics. (2008). Edinburgh: Churchill Livingstone Elsevier.
Pisters, P. W., O'Sullivan, B., & Maki, R. G. (2007). Evidence-based recommendations for local therapy for soft tissue sarcomas. J Clin Oncol, 25(8), 1003-1008.
Pisters, P. W. T. (2002). Retroperitoneal sarcomas. I: R. E. Pollock (red.), Soft tissue sarcomas (s. 180-188). Hamilton: Decker.
Pårørendeveileder. (28.01.2019). [nettdokument]. Oslo: Helsedirektoratet. Hentet 31.01.2022, fra https://www.helsedirektoratet.no/veiledere/parorendeveileder
Rakheja, R., Makis, W., Skamene, S., Nahal, A., Brimo, F., Azoulay, L., . . . Hickeson, M. (2012). Correlating metabolic activity on 18F-FDG PET/CT with histopathologic characteristics of osseous and soft-tissue sarcomas: a retrospective review of 136 patients. AJR Am J Roentgenol, 198(6), 1409-1416. https://doi.org/10.2214/AJR.11.7560
Riedel, R. F., Larrier, N., Dodd, L., Kirsch, D., Martinez, S., & Brigman, B. E. (2009). The clinical management of chondrosarcoma. Curr Treat Options Oncol, 10(1-2), 94-106.
Roberge, D., Vakilian, S., Alabed, Y. Z., Turcotte, R. E., Freeman, C. R., & Hickeson, M. (2012). FDG PET/CT in Initial Staging of Adult Soft-Tissue Sarcoma. Sarcoma, 2012, 960194. https://doi.org/10.1155/2012/960194
Robertson, E. G., & Baxter, G. (2011). Tumour seeding following percutaneous needle biopsy: the real story! Clinical Radiology, 66(11), 1007-1014. https://doi.org/10.1016/j.crad.2011.05.012
Rombi, B., DeLaney, T. F., MacDonald, S. M., Huang, M. S., Ebb, D. H., Liebsch, N. J., . . . Yock, T. I. (2012). Proton radiotherapy for pediatric Ewing's sarcoma: initial clinical outcomes. Int J Radiat Oncol Biol Phys, 82(3), 1142-1148. https://doi.org/10.1016/j.ijrobp.2011.03.038
Rosenberg, A. E., Nielsen, G. P., Keel, S. B., Renard, L. G., Fitzek, M. M., Munzenrider, J. E., & Liebsch, N. J. (1999). Chondrosarcoma of the base of the skull: a clinicopathologic study of 200 cases with emphasis on its distinction from chordoma. Am J Surg Pathol, 23(11), 1370-1378.
Rowbotham, E., Bhuva, S., Gupta, H., & Robinson, P. (2012). Assessment of referrals into the soft tissue sarcoma service: evaluation of imaging early in the pathway process. Sarcoma, 2012, 781723. https://doi.org/10.1155/2012/781723
Rydholm, A. (1998). Improving the management of soft tissue sarcoma. Diagnosis and treatment should be given in specialist centres. BMJ, 317(7151), 93-94.
Salunke, A. A., Chen, Y., Tan, J. H., Chen, X., Khin, L. W., & Puhaindran, M. E. (2014). Does a pathological fracture affect the prognosis in patients with osteosarcoma of the extremities? : a systematic review and meta-analysis. The bone & joint journal, 96-B(10), 1396-1403. https://doi.org/10.1302/0301-620X.96B10.34370
Sarkom. (23.08. 2018 ). I: Pakkeforløp for kreft - Diagnoseveiledere. [nettdokument]. Oslo: Helsedirektoratet. Hentet 31.01.2022
Schmidt-Braekling, T., Streitbuerger, A., Gosheger, G., Boettner, F., Nottrott, M., Ahrens, H., . . . Hardes, J. (2017). Silver-coated megaprostheses: review of the literature. European journal of orthopaedic surgery & traumatology : orthopedie traumatologie, 27(4), 483-489. https://doi.org/10.1007/s00590-017-1933-9
Schwartz, H. S., Zimmerman, N. B., Simon, M. A., Wroble, R. R., Millar, E. A., & Bonfiglio, M. (1987). The malignant potential of enchondromatosis. J Bone Joint Surg Am, 69(2), 269-274.
Schöffski, P., Chawla, S., Maki, R. G., Italiano, A., Gelderblom, H., Choy, E., . . . Patel, S. R. (2016). Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet, 387(10028), 1629-1637. https://doi.org/10.1016/s0140-6736(15)01283-0
Seddon, B., Strauss, S. J., Whelan, J., Leahy, M., Woll, P. J., Cowie, F., . . . Beare, S. (2017). Gemcitabine and docetaxel versus doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft-tissue sarcomas (GeDDiS): a randomised controlled phase 3 trial. The Lancet. Oncology, Sep 4 [Epub ahead of print]. https://doi.org/10.1016/s1470-2045(17)30622-8
Seneffekter etter kreftbehandling. (2020). (IS-2872). Oslo: Helsedirektoratet. Hentet fra https://www.helsedirektoratet.no/rapporter/seneffekter-etter-kreftbehandling/Seneffekter%20etter%20kreftbehandling.pdf/_/attachment/inline/3d984c2a-7926-4d1a-a5f0-06d48fe7c95f:f3e498d059734ff34b013c1c206877e488e95600/Seneffekter%20etter%20kreftbehandling.pdf
Sequential Neoadjuvant Chemotherapy in Soft Tissue Sarcoma. (2021-2034). [pågående studie]. (NCT04776525). Hentet fra https://clinicaltrials.gov/ct2/show/NCT04776525
Sleijfer, S., Ray-Coquard, I., Papai, Z., Le Cesne, A., Scurr, M., Schoffski, P., . . . Blay, J. Y. (2009). Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043). J Clin Oncol, 27(19), 3126-3132. https://doi.org/10.1200/jco.2008.21.3223
Smith, H. G., Cartwright, J., Wilkinson, M. J., Strauss, D. C., Thomas, J. M., & Hayes, A. J. (2015). Isolated Limb Perfusion with Melphalan and Tumour Necrosis Factor alpha for In-Transit Melanoma and Soft Tissue Sarcoma. Annals of Surgical Oncology, 22 Suppl 3, S356-361.
Smith, L. M., & Donaldson, S. S. (1991). Incidence and management of secondary malignancies in patients with retinoblastoma and Ewing's sarcoma. Oncology (Williston Park, N.Y.), 5(5), 135-141; discussion 142.
Smith, M. J., Ridgway, P. F., Catton, C. N., Cannell, A. J., O'Sullivan, B., Mikula, L. A., . . . Swallow, C. J. (2014). Combined management of retroperitoneal sarcoma with dose intensification radiotherapy and resection: long-term results of a prospective trial. Radiother Oncol, 110(1), 165-171. https://doi.org/10.1016/j.radonc.2013.10.041
Souba, W. W., McKenna, R. J., Jr., Meis, J., Benjamin, R., Raymond, A. K., & Mountain, C. F. (1986). Radiation-induced sarcomas of the chest wall. Cancer, 57(3), 610-615.
SSG XXV: The Stop-GIST Trial; Discontinuation of Imatinib in Patients With Oligo-metastatic GIST. (2016-2022). [pågående studie]. (NCT02924714). Hentet fra https://clinicaltrials.gov/ct2/show/NCT02924714
Stacchiotti, S., Tortoreto, M., Bozzi, F., Tamborini, E., Morosi, C., Messina, A., . . . Casali, P. G. (2013). Dacarbazine in solitary fibrous tumor: a case series analysis and preclinical evidence vis-a-vis temozolomide and antiangiogenics. Clinical cancer research : an official journal of the American Association for Cancer Research, 19(18), 5192-5201. https://doi.org/10.1158/1078-0432.Ccr-13-0776
Stoeckle, E., Coindre, J. M., Bonvalot, S., Kantor, G., Terrier, P., Bonichon, F., . . . French Federation of Cancer Centers Sarcoma, G. (2001). Prognostic factors in retroperitoneal sarcoma: a multivariate analysis of a series of 165 patients of the French Cancer Center Federation Sarcoma Group. Cancer, 92(2), 359-368.
Stoldt, H. S., & Geraghty, J. G. (1999). Surgical principles in the management of soft tissue sarcomas. I: I. Taylor & C. D. Johnson (red.), Recent advances in surgery (s. 47-61). London: Royal Society of Medicine Press.
Strander, H., Bauer, H. C., Brosjö, O., Fernberg, J. O., Kreicbergs, A., Nilsonne, U., . . . Söderlund, V. (1995). Long-term adjuvant interferon treatment of human osteosarcoma. A pilot study. Acta oncologica (Stockholm, Sweden), 34(6), 877-880.
Strander, H., & Einhorn, S. (1977). Effect of human leukocyte interferon on the growth of human osteosarcoma cells in tissue culture. Int J Cancer, 19(4), 468-473.
Strauss, S. J., Frezza, A. M., Abecassis, N., Bajpai, J., Bauer, S., Biagini, R., . . . Stacchiotti, S. (2021). Bone sarcomas: ESMO-EURACAN-GENTURIS-ERN PaedCan Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol, 32(12), 1520-1536. https://doi.org/10.1016/j.annonc.2021.08.1995
Suit, H., Kooy, H., Trofimov, A., Farr, J., Munzenrider, J., DeLaney, T., . . . Paganetti, H. (2008). Should positive phase III clinical trial data be required before proton beam therapy is more widely adopted? No. Radiother Oncol, 86(2), 148-153. https://doi.org/10.1016/j.radonc.2007.12.024
Sundby Hall, K., Bruland Ø, S., Bjerkehagen, B., Zaikova, O., Engellau, J., Hagberg, O., . . . Eriksson, M. (2018). Adjuvant chemotherapy and postoperative radiotherapy in high-risk soft tissue sarcoma patients defined by biological risk factors-A Scandinavian Sarcoma Group study (SSG XX). Eur J Cancer, 99, 78-85. https://doi.org/10.1016/j.ejca.2018.05.011
Surgery With Our Without Neoadjuvant Chemotherapy in High Risk RetroPeritoneal Sarcoma (STRASS2). (2019-2028). [pågående studie]. (NCT04031677). Hentet fra https://clinicaltrials.gov/ct2/show/NCT04031677
Swanson, E. L., Indelicato, D. J., Louis, D., Flampouri, S., Li, Z., Morris, C. G., . . . Slopsema, R. (2012). Comparison of three-dimensional (3D) conformal proton radiotherapy (RT), 3D conformal photon RT, and intensity-modulated RT for retroperitoneal and intra-abdominal sarcomas. Int J Radiat Oncol Biol Phys, 83(5), 1549-1557. https://doi.org/10.1016/j.ijrobp.2011.10.014
Tangvik, R. J., Tell, G. S., Guttormsen, A. B., Eisman, J. A., Henriksen, A., Nilsen, R. M., & Ranhoff, A. H. (2015). Nutritional risk profile in a university hospital population. Clinical Nutrition, 34(4), 705-711.
Tawbi, H. A., Burgess, M., Bolejack, V., Van Tine, B. A., Schuetze, S. M., Hu, J., . . . Patel, S. (2017). Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. The Lancet. Oncology, 18(11), 1493-1501. https://doi.org/10.1016/s1470-2045(17)30624-1
Tehranzadeh, J., Mnaymneh, W., Ghavam, C., Morillo, G., & Murphy, B. J. (1989). Comparison of CT and MR imaging in musculoskeletal neoplasms. J Comput Assist Tomogr, 13(3), 466-472.
Thomas, H., & Timmermann, B. (2020). Paediatric proton therapy. British Journal of Radiology, 93(1107), 20190601. https://doi.org/10.1259/bjr.20190601
Thoresen, S. O. (1992). Li-Fraumeni-syndromet og p53-genet. Tidsskr Nor Lægeforen, 112(7), 887-889.
Toguchida, J., Yamaguchi, T., Dayton, S. H., Beauchamp, R. L., Herrera, G. E., Ishizaki, K., . . . Sasaki, M. S. (1992). Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. New England Journal of Medicine, 326(20), 1301-1308.
Toulmonde, M., Bonvalot, S., Méeus, P., Stoeckle, E., Riou, O., Isambert, N., . . . Italiano, A. (2014). Retroperitoneal sarcomas: patterns of care at diagnosis, prognostic factors and focus on main histological subtypes: a multicenter analysis of the French Sarcoma Group. Ann Oncol, 25(3), 735-742. https://doi.org/10.1093/annonc/mdt577
Travis, L. B., Rabkin, C. S., Brown, L. M., Allan, J. M., Alter, B. P., Ambrosone, C. B., . . . Greene, M. H. (2006). Cancer survivorship--genetic susceptibility and second primary cancers: research strategies and recommendations. J Natl Cancer Inst, 98(1), 15-25.
Trovik, C., Bauer, H. C. F., Styring, E., Sundby Hall, K., Vult Von Steyern, F., Eriksson, S., . . . Alvegard, T. A. (2017). The Scandinavian Sarcoma Group Central Register: 6,000 patients after 25 years of monitoring of referral and treatment of extremity and trunk wall soft-tissue sarcoma. Acta orthopaedica, 88(3), 341-347. https://doi.org/10.1080/17453674.2017.1293441
Trovik, C. S., Bauer, H. C., Berlin, O., Tukiainen, E., Erlanson, M., Gustafson, P., . . . Wahlstrom, O. (2001). Local recurrence of deep-seated, high-grade, soft tissue sarcoma: 459 patients from the Scandinavian Sarcoma Group Register. Acta Orthop Scand, 72(2), 160-166.
Trovik, L. H., Ovrebo, K., Almquist, M., Haugland, H. K., Rissler, P., Eide, J., . . . Jebsen, N. L. (2014). Adjuvant radiotherapy in retroperitoneal sarcomas. A Scandinavian Sarcoma Group study of 97 patients. Acta Oncologica, 53(9), 1165-1172.
Tsuchiya, T., Sekine, K., Hinohara, S., Namiki, T., Nobori, T., & Kaneko, Y. (2000). Analysis of the p16INK4, p14ARF, p15, TP53, and MDM2 genes and their prognostic implications in osteosarcoma and Ewing sarcoma. Cancer Genet Cytogenet, 120(2), 91-98.
Uhl, M., Edler, L., Jensen, A. D., Habl, G., Oelmann, J., Röder, F., . . . Herfarth, K. (2014). Randomized phase II trial of hypofractionated proton versus carbon ion radiation therapy in patients with sacrococcygeal chordoma-the ISAC trial protocol. Radiation oncology, 9, 100. https://doi.org/10.1186/1748-717X-9-100
Upubliserte data.
van der Graaf, W. T., Blay, J. Y., Chawla, S. P., Kim, D. W., Bui-Nguyen, B., Casali, P. G., . . . Hohenberger, P. (2012). Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet, 379(9829), 1879-1886. https://doi.org/10.1016/s0140-6736(12)60651-5
Van Glabbeke, M., Verweij, J., Judson, I., & Nielsen, O. S. (2002). Progression-free rate as the principal end-point for phase II trials in soft-tissue sarcomas. Eur J Cancer, 38(4), 543-549. https://doi.org/10.1016/s0959-8049(01)00398-7
van Houdt, W. J., NS, I. J., Marjolein Schrijver, A., Huis In 't Veld, E., Thway, K., Jones, R. L., . . . Smith, M. J. F. (2021). Oncological Outcome After Diagnostic Biopsies in Gastrointestinal Stromal Tumors: A Retrospective Cohort Study. Ann Surg, 274(6), e1093-e1098. https://doi.org/10.1097/sla.0000000000003744
van Houdt, W. J., Raut, C. P., Bonvalot, S., Swallow, C. J., Haas, R., & Gronchi, A. (2019). New research strategies in retroperitoneal sarcoma. The case of TARPSWG, STRASS and RESAR: making progress through collaboration. Current Opinion in Oncology, 31(4), 310-316. https://doi.org/10.1097/cco.0000000000000535
Van Winkle, P., Angiolillo, A., Krailo, M., Cheung, Y. K., Anderson, B., Davenport, V., . . . Cairo, M. S. (2005). Ifosfamide, carboplatin, and etoposide (ICE) reinduction chemotherapy in a large cohort of children and adolescents with recurrent/refractory sarcoma: the Children's Cancer Group (CCG) experience. Pediatric blood & cancer, 44(4), 338-347. https://doi.org/10.1002/pbc.20227
Vincenzi, B., Napolitano, A., Fiocco, M., Mir, O., Rutkowski, P., Blay, J. Y., . . . Casali, P. G. (2021). Adjuvant Imatinib in Patients with GIST Harboring Exon 9 KIT Mutations: Results from a Multi-institutional European Retrospective Study. Clinical cancer research : an official journal of the American Association for Cancer Research. https://doi.org/10.1158/1078-0432.Ccr-21-1665
Wafa, H., Grimer, R. J., Reddy, K., Jeys, L., Abudu, A., Carter, S. R., & Tillman, R. M. (2015). Retrospective evaluation of the incidence of early periprosthetic infection with silver-treated endoprostheses in high-risk patients: case-control study. The bone & joint journal, 97-B(2), 252-257. https://doi.org/10.1302/0301-620X.97B2.34554
Wang, D., Bosch, W., Roberge, D., Finkelstein, S. E., Petersen, I., Haddock, M., . . . DeLaney, T. F. (2011). RTOG sarcoma radiation oncologists reach consensus on gross tumor volume and clinical target volume on computed tomographic images for preoperative radiotherapy of primary soft tissue sarcoma of extremity in Radiation Therapy Oncology Group studies. Int J Radiat Oncol Biol Phys, 81(4), e525-528. https://doi.org/10.1016/j.ijrobp.2011.04.038
Weatherall, P. T. (1995). Benign and malignant masses. MR imaging differentiation. Magn Reson Imaging Clin N Am, 3(4), 669-694.
Weber, D. C., Murray, F. R., Correia, D., Bolsi, A., Frei-Welte, M., Pica, A., . . . Bachtiary, B. (2017). Pencil beam scanned protons for the treatment of patients with Ewing sarcoma. Pediatric blood & cancer, 64(12). https://doi.org/10.1002/pbc.26688
Weber, D. C., Trofimov, A. V., Delaney, T. F., & Bortfeld, T. (2004). A treatment planning comparison of intensity modulated photon and proton therapy for paraspinal sarcomas. Int J Radiat Oncol Biol Phys, 58(5), 1596-1606. https://doi.org/10.1016/j.ijrobp.2003.11.028
Whelan, J., Le Deley, M. C., Dirksen, U., Le Teuff, G., Brennan, B., Gaspar, N., . . . Oberlin, O. (2018). High-Dose Chemotherapy and Blood Autologous Stem-Cell Rescue Compared With Standard Chemotherapy in Localized High-Risk Ewing Sarcoma: Results of Euro-E.W.I.N.G.99 and Ewing-2008. J Clin Oncol, 36(31), Jco2018782516. https://doi.org/10.1200/jco.2018.78.2516
WHO Classification of Tumours Editorial Board (red.). (2020). WHO Classification of Tumours of Soft Tissue and Bone (5. utg.). (WHO Classification of Tumours, Volume 3). Geneva: WHO.
Wick, M. R., Siegal, G. P., Unni, K. K., McLeod, R. A., & Greditzer, H. G., III. (1981). Sarcomas of bone complicating osteitis deformans (Paget's disease): fifty years' experience. Am J Surg Pathol, 5(1), 47-59.
Widhe, B., & Widhe, T. (2000). Initial symptoms and clinical features in osteosarcoma and Ewing sarcoma. J Bone Joint Surg Am, 82(5), 667-674.
Wilky, B. A., Trucco, M. M., Subhawong, T. K., Florou, V., Park, W., Kwon, D., . . . Trent, J. C. (2019). Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: a single-centre, single-arm, phase 2 trial. The Lancet. Oncology, 20(6), 837-848. https://doi.org/10.1016/s1470-2045(19)30153-6
Wittekind, C., Compton, C. C., Greene, F. L., & Sobin, L. H. (2002). TNM residual tumor classification revisited. Cancer, 94(9), 2511-2516.
Wolfson, A. H., Benedetto, P. W., Mnaymneh, W., Moffat, F. L., Robinson, D. S., Boyer, C., . . . Markoe, A. M. (1998). Does a radiation dose-response relation exist concerning survival of patients who have soft-tissue sarcomas of the extremities? Radiation dose-response relation for soft-tissue sarcomas. Am J Clin Oncol, 21(3), 270-274.
Woll, P. J., Reichardt, P., Le Cesne, A., Bonvalot, S., Azzarelli, A., Hoekstra, H. J., . . . the, N. C. T. G. S. D. S. C. (2012). Adjuvant chemotherapy with doxorubicin, ifosfamide, and lenograstim for resected soft-tissue sarcoma (EORTC 62931): a multicentre randomised controlled trial. Lancet Oncology, 13(10), 1045-1054.
Yang, J. C., Chang, A. E., Baker, A. R., Sindelar, W. F., Danforth, D. N., Topalian, S. L., . . . Rosenberg, S. A. (1998). Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol, 16(1), 197-203.
Yap, J., Chuba, P. J., Thomas, R., Aref, A., Lucas, D., Severson, R. K., & Hamre, M. (2002). Sarcoma as a second malignancy after treatment for breast cancer. Int J Radiat Oncol Biol Phys, 52(5), 1231-1237.
Zagars, G. K., & Ballo, M. T. (2003). Significance of dose in postoperative radiotherapy for soft tissue sarcoma. Int J Radiat Oncol Biol Phys, 56(2), 473-481.
Zaikova, O., Sundby Hall, K., Styring, E., Eriksson, M., Trovik, C. S., Bergh, P., . . . Bauer, H. C. (2015). Referral patterns, treatment and outcome of high-grade malignant bone sarcoma in Scandinavia--SSG Central Register 25 years' experience. Journal of Surgical Oncology, 112(8), 853-860.
Zalcberg, J. R., Verweij, J., Casali, P. G., Le Cesne, A., Reichardt, P., Blay, J. Y., . . . Australasian Gastrointestinal Trials, G. (2005). Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. European Journal of Cancer, 41(12), 1751-1757.
Zhou, H., Coffin, C. M., Perkins, S. L., Tripp, S. R., Liew, M., & Viskochil, D. H. (2003). Malignant peripheral nerve sheath tumor: a comparison of grade, immunophenotype, and cell cycle/growth activation marker expression in sporadic and neurofibromatosis 1-related lesions. Am J Surg Pathol, 27(10), 1337-1345.
Utgåtte versjoner
Sist faglig oppdatert: 28.02.2022