









Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av kreft i magesekken (ventrikkelkreft)
Forord
Sist faglig oppdatert: 28.09.2021
Nasjonale handlingsprogram med faglige retningslinjer for kreft skal bidra til at det offentlige tilbudet i kreftomsorgen blir av god kvalitet og likeverdig over hele landet. Målgrupper for retningslinjene er leger og legespesialister innen medisin, kirurgi, onkologi, radiologi, patologi og fastleger. De vil også være av interesse for andre faggrupper som er involvert i behandling og oppfølging av kreftpasienter og deres pårørende.
Nasjonale faglige retningslinjer fra Helsedirektoratet er å betrakte som anbefalinger og råd, basert på oppdatert faglig kunnskap. De nasjonale faglige retningslinjene gir uttrykk for hva som anses som god praksis på utgivelsestidspunktet, og er ment som et hjelpemiddel ved de avveininger tjenesteyterne må gjøre for å oppnå forvarlighet og god kvalitet i tjenesten. Nasjonale faglige retningslinjer er ikke direkte rettslig bindende for mottagerne, men bør langt på vei være styrende for de valg som skal tas. Ved å følge oppdaterte nasjonale faglige retningslinjer, vil fagpersonell bidra til å oppfylle kravet om faglig forsvarlighet. Dersom en velger løsninger som i vesentlig grad avviker fra de nasjonale faglige retningslinjene, bør en dokumentere dette, og være forberedt på å begrunne sine valg. Sykehusenes eiere og ledelse bør tilrettelegge virksomheten slik at de nasjonale faglige retningslinjene kan følges.
Helsedirektoratet takker arbeidsgruppen for stor innsats i utarbeidelsen av handlingsprogrammet. Vi håper handlingsprogrammet vil være et nyttig arbeidsredskap ved behandling av pasienter med magekreft. Innholdet i den nasjonale retningslinjen for magekreft vil vurderes årlig, og om nødvendig oppdateres.
Disse nasjonale faglige retningslinjene for diagnostikk, behandling og oppfølging av pasienter med magekreft er publisert 28. september 2021.
Bjørn Guldvog
Helsedirektør
Sammendrag av retningslinjene
Sist faglig oppdatert: 28.09.2021
| Retningslinjer | Kunnskapsgrunnlagets evidensgrad |
|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Kirurgisk behandling |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Innledning
Sist faglig oppdatert: 28.09.2021
Endringer i foreliggende 5. utgave av handlingsprogrammet utgitt 15.06.2018
Nytt kapittel om arvelig kreft i magesekk:
Diagnose og utredning
- Ny tekst om diagnostisk laparoskopi
- Ny TNM-klassifikasjon er lagt inn, inkludert en klassifikasjon for klinisk stadium
Behandling av lokalisert sykdom
- Delkapittelet om medikamentell behandling er skrevet om
Behandling av metastaserende sykdom
- Nytt kapittel om kirurgi som livsforlengende behandling ved langt fremskreden sykdom
- Delkapittelet om medikamentell behandling er skrevet om
Epidemiologi
Forekomst
Sist faglig oppdatert: 28.09.2021
Det oppdages omtrent 400 nye tilfeller med kreft i magesekken i Norge årlig (2018), og frekvensen er fortsatt avtagende. For om lag 20 år siden var det 1100 nye tilfeller årlig. Gjennomsnittlig alder ved diagnose er 75 år, og 60 % er menn (Larsen, 2016). Det er i hovedsak to ulike typer kreft i magesekken: intestinal og diffus type (Lauréns klassifikasjon (Lauren, 1965)), og det er vesentlig den intestinale formen som avtar i hyppighet.
Kjente risikofaktorer er infeksjon med Helicobacter pylori, røyking og tidligere gjennomgått Bilroth II-reseksjon for ulcus pepticum (Forman & Burley, 2006; Larsson, Bergkvist, & Wolk, 2006) (evidensnivå 2a).
Diffus type er muligens relatert til genetisk etiologi samt infeksjon med Epstein-Barr virus. Prognosen ved diffus type er dårligere enn ved intestinal type (Viste, Eide, Halvorsen, Maartmann-Moe, & Søreide, 1986).
På diagnosetidspunkt er 18 % (både menn og kvinner) av svulstene lokalisert kun til veggen i magesekken, 30 % av mennene og 24 % av kvinnene har lymfeknutemetastaser og 31 % (både menn og kvinner) har fjernmetastaser.
Insidens (forekomst), mortalitet (dødelighet) og relativ overlevelse av kreft i magesekken i Norge for kvinner og menn:
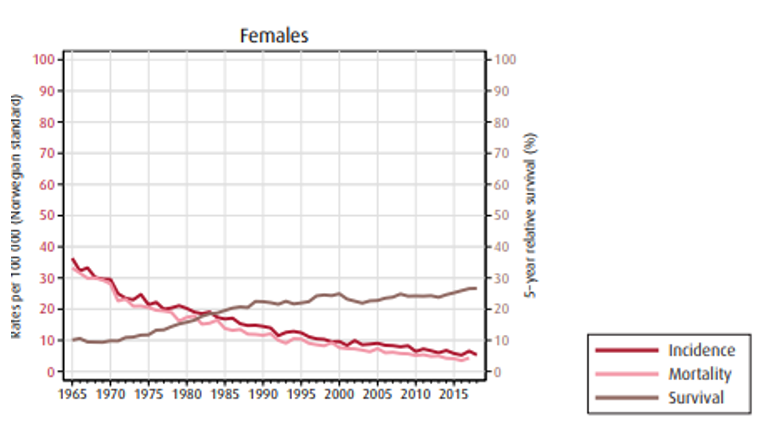
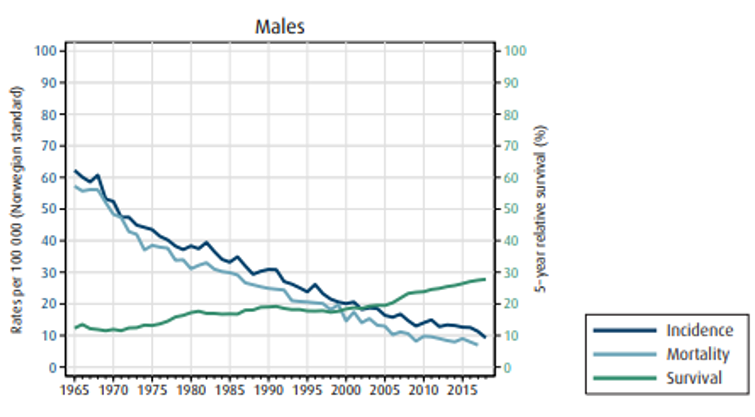
Overlevelse
Sist faglig oppdatert: 28.09.2021
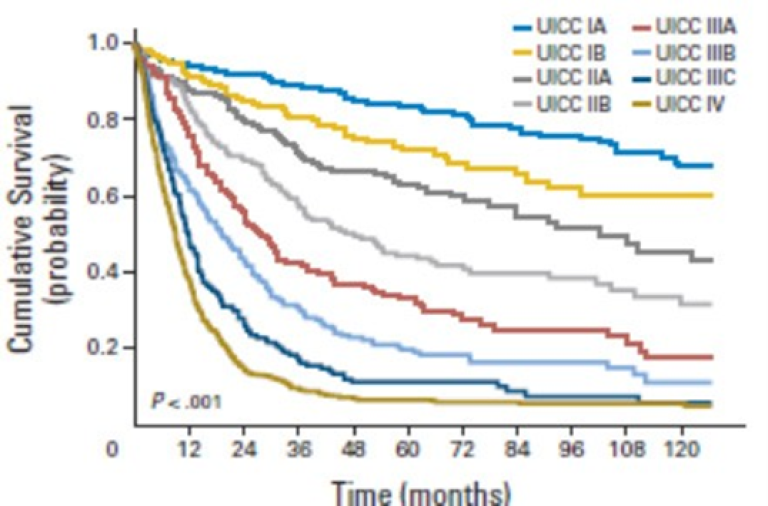
Overlevelse ved kreft i magesekken har for hele pasientgruppen økt fra 17 % i perioden 1976- 1980 til 27 % i perioden 2013- 2017 for menn og fra 15 % til 26 % for kvinner. Overlevelse er sterkt relatert til sykdomsstadium med 60 % fem-års overlevelse for lokalisert sykdom for menn (58 % for kvinner), mens kun 3 % av pasienter (både menn og kvinner) med fjernspredning er i live etter 5 år .
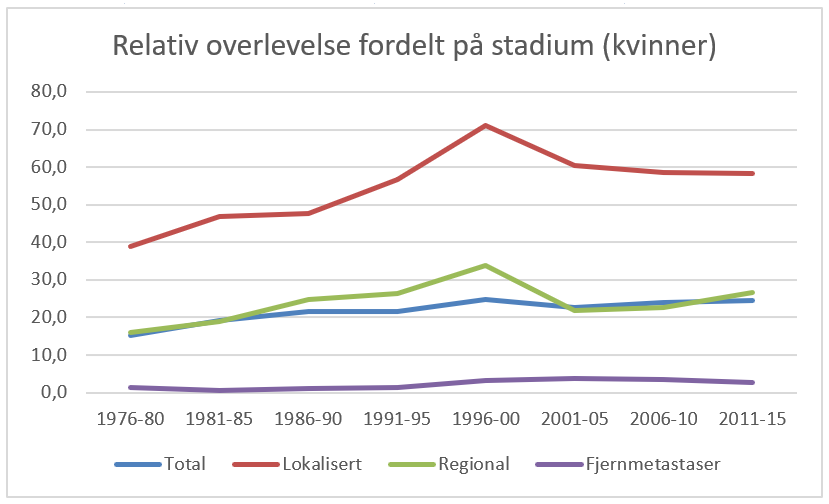
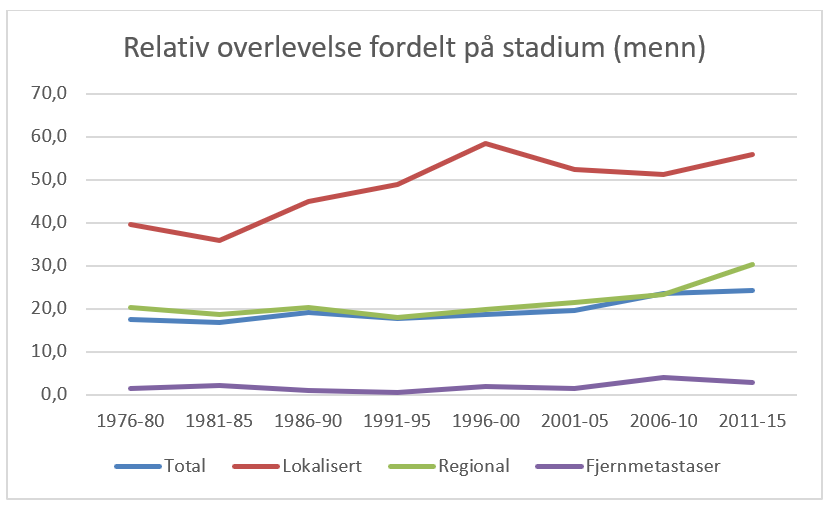
Forebygging
Sist faglig oppdatert: 28.09.2021
Helicobacter pylori er på verdensbasis en viktig risikofaktor. Høy prevalens er assosiert med høy forekomst av kreft i magesekken. Kostholdets betydning som risikofaktor er kompleks. Generelt er inntak av planteprodukter relatert til redusert risiko for kreft, mens høyt inntak av animalske produkter er relatert til økt risiko (Mayne et al., 2001). Kosthold med høyt innhold av antioksidanter som vitamin A (retinol og betakarotener), vitamin C, vitamin E er assosiert med redusert risiko for kreft i magesekken (Kong et al., 2014) (evidensnivå 1a). Livsstilsfaktorer som kosthold, røyk og alkohol påvirker også risiko (den Hoed & Kuipers, 2016). En sunn livsstilsindeks ble definert og undersøkt i den store europeiske EPIC studien med 500.000 deltakere.Indeksen ble satt sammen av røyking, alkohol, kostfaktorer og vekt. En høy score på definert livsstilsindeks var knyttet til 50 % reduksjon av kreft i magesekk (Buckland et al., 2015). Middelhavskosten var foretrukket. Ytterligere faktorer i kosten som øker risiko for kreft er bl.a. høyt innhold av salt og syltet, røkt eller stekt mat (Yusefi, Bagheri Lankarani, Bastani, Radinmanesh, & Kavosi, 2018).
Arvelig kreft i magesekken
Sist faglig oppdatert: 28.09.2021
Pasienter med forskjellige syndromer som hovedsakelig domineres av økt forekomst av polypper, har også en arvelig predisposisjon for kreft i magesekken. Eksempler er syndromer som Li-Fraumeni, Lynch, Peutz- Jegher og juvenil polypose samt ved arvelig brystkreft og eggstokk-kreft, familiær adenomatøs polypose og hamartom tumor syndrom (Cowden syndrom). Livstidsrisikoen for kreft i magesekken i disse syndromene varierer, men er generelt forholdsvis lav.
Arvelig diffus kreft i magesekken
Sist faglig oppdatert: 28.09.2021
1-3 % av pasientene med kreft i magesekken har arvelig diffus kreft i magesekken (heriditær diffuse gastric cancer = HDGC). Ved denne tilstanden foreligger mutasjon i tumor suppressor genet CDH1, som koder for celle-adhesjons-proteinet E-cadherin. Arvegangen er autosomal dominant med mer enn 80% penetrasjon. 25-50 % av pasientene som oppfyller kriteriene på HDGC har kimcellemutasjoner i dette genet (Fitzgerald et al., 2010). Gjennomsnittsalder ved diagnose av denne type arvelig kreft i magesekken er 38 år. Livstidsrisikoen hos disse pasientene for å utvikle kreft i magesekken ved alder 80 år er rapportert til 70 % (95 % konfidensintervall 59-80 %) for menn og 56 % for kvinner (95 % konfidensintervall 23-68 %) (van der Post et al., 2015). Kreft i magesekken av diffus type foreligger vanligvis i tumor stadium T1N0. I tillegg er livstidsrisiko for å utvikle brystkreft av lobulær type blant kvinner med CDH1 mutasjon 40-50 %.
Diagnose
Ett av følgende tre diagnostiske kriterier må foreligge i 1. og/ eller 2. grads slektninger for å kunne identifisere pasienter med HDGC:
- Minst to pasienter med kreft i magesekken uansett alder, minst én med kreft i magesekken av diffus type
- En pasient med diffus kreft i magesekken ved alder < 40 år
- Personlig eller familiehistorie med kreft i magesekken av diffus type og dessuten lobulær brystkreft, hvorav et tilfelle ved alder < 50 år
Pasienter med kreftsykdommen vil bli undersøkt og hvis der foreligger sykdomsfremkallende CDH1 mutasjon, anbefales alle familiemedlemmer fra alder 16 år og oppover å bli undersøkt med henblikk på profylaktisk behandling.
Behandling og overvåking
Profylaktisk total gastrektomi hos individer med påvist sykdomsfremkallende CDH1-mutasjon tilrådes fra 18-20-årsalderen. Gastroskopisk overvåking med multiple biopsier (>30) er ikke tilstrekkelig for å utelukke tidlig kreftsykdom, men må gjøres hvert år hvis pasienten ikke ønsker profylaktisk gastrektomi. Multiple tidlige signetringcellecancer foci (T1N0) er påvist hos 100 % av profylaktiske gastrektomier hos CDH1-positive pasienter. Kvinner med CDH1-mutasjon anbefales hvert år mammografi og MR av brystene Mutasjonsundersøkelse er kun positiv i 25-50 % av familiene. I CDH1-negative familier anbefales årlig gastroskopi med multiple biopsier og for kvinner mammografi og MR av brystene.
Gastrisk adenokarsinom og proksimal polypose i magesekken
Sist faglig oppdatert: 28.09.2021
Gastrisk adenokarsinom og proksimal polypose i magesekken eller gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS) er en meget sjelden arvelig variant av kreft i magesekken første gang beskrevet i 2012 (Rudloff, 2018). Polypose syndromet med risiko for utvikling av kreft i magesekken ble påvist i en stor familie i Australia og i to mindre familier I USA og Canada. Prevalens av dette syndromet er ukjent, men GAPPS er også påvist i Norge. Ved denne tilstanden ble det i 2016 beskrevet mutasjoner i det såkalte promotor 1B adenomatøs polyposis coli (APC) genet som hemmer genets transkripsjon. Arvegangen er autosomal dominant, men penetransen er sannsynlig ufullstendig siden noen genbærere kan ha normale endoskopifunn gjennom hele livet. GAPPS er karakterisert ved utvikling av 100 polypper eller flere i fundus- og corpusdelen av magesekken, men ikke i antrum. Karakteristiske endoskopiske funn er polypper av varierende størrelse, vanligvis mindre enn 10 mm, og kan være tilstede fra alder rundt 10 år. Tidligste mikroskopiske tegn synes å være hyperproliferative ansamlinger av spesialiserte oxyntiske celler i mucosa (de Boer, Ee, & Kumarasinghe, 2018). Pasientene med GAPPS som utvikler adenokarsinom i dysplastiske polypper er ofte unge i alder 20- og 30-årene (van der Post, Oliveira, Guilford, & Carneiro, 2019).
Diagnose
Følgende kriterier må være oppfylt for å stille diagnosen:
- Polypper avgrenset til fundus- og corpus, uten funn av polypper i duodenum, colon og rectum
- Mer enn 100 polypper hos indeks pasient, eller mer enn 30 polypper hos 1. grads slektning
- Hovedsakelig funn av funduspolypper, men noen har også funn av dysplasi eller et familiemedlem med enten dysplastiske funduspolypper eller adenokarsinom i magesekken
- Autosomal dominant arvegang
- Utelukke andre arvelige gastriske polypose syndromer og bruk av protonpumpehemmere
- Påvisning av mutasjonen i APC genet
Behandling og overvåking
Det finnes ingen konsensus på hvordan disse pasientene bør følges opp eller når man eventuelt skal tilby gastrektomi (Rudloff, 2018). Oppfølging med CT thorax/ abdomen/bekken og CT-ventriculografi, i tillegg til regelmessige gastroskopier og multiple biopsier, må balanseres mot risiko for utvikling av kreft og behov for behandling med gastrektomi. Endoskopisk oppfølging har begrensinger siden utvikling av tidlig stadium av kraft har vært oversett med påfølgende utvikling av metastaserende sykdom.
Arvelig diffus kreft i magesekken
Sist faglig oppdatert: 28.09.2021
1-3 % av pasientene med kreft i magesekken har arvelig diffus kreft i magesekken (heriditær diffuse gastric cancer = HDGC). Ved denne tilstanden foreligger mutasjon i tumor suppressor genet CDH1, som koder for celle-adhesjons-proteinet E-cadherin. Arvegangen er autosomal dominant med mer enn 80% penetrasjon. 25-50 % av pasientene som oppfyller kriteriene på HDGC har kimcellemutasjoner i dette genet (Fitzgerald et al., 2010). Gjennomsnittsalder ved diagnose av denne type arvelig kreft i magesekken er 38 år. Livstidsrisikoen hos disse pasientene for å utvikle kreft i magesekken ved alder 80 år er rapportert til 70 % (95 % konfidensintervall 59-80 %) for menn og 56 % for kvinner (95 % konfidensintervall 23-68 %) (van der Post et al., 2015). Kreft i magesekken av diffus type foreligger vanligvis i tumor stadium T1N0. I tillegg er livstidsrisiko for å utvikle brystkreft av lobulær type blant kvinner med CDH1 mutasjon 40-50 %.
Diagnose
Ett av følgende tre diagnostiske kriterier må foreligge i 1. og/ eller 2. grads slektninger for å kunne identifisere pasienter med HDGC:
- Minst to pasienter med kreft i magesekken uansett alder, minst én med kreft i magesekken av diffus type
- En pasient med diffus kreft i magesekken ved alder < 40 år
- Personlig eller familiehistorie med kreft i magesekken av diffus type og dessuten lobulær brystkreft, hvorav et tilfelle ved alder < 50 år
Pasienter med kreftsykdommen vil bli undersøkt og hvis der foreligger sykdomsfremkallende CDH1 mutasjon, anbefales alle familiemedlemmer fra alder 16 år og oppover å bli undersøkt med henblikk på profylaktisk behandling.
Behandling og overvåking
Profylaktisk total gastrektomi hos individer med påvist sykdomsfremkallende CDH1-mutasjon tilrådes fra 18-20-årsalderen. Gastroskopisk overvåking med multiple biopsier (>30) er ikke tilstrekkelig for å utelukke tidlig kreftsykdom, men må gjøres hvert år hvis pasienten ikke ønsker profylaktisk gastrektomi. Multiple tidlige signetringcellecancer foci (T1N0) er påvist hos 100 % av profylaktiske gastrektomier hos CDH1-positive pasienter. Kvinner med CDH1-mutasjon anbefales hvert år mammografi og MR av brystene Mutasjonsundersøkelse er kun positiv i 25-50 % av familiene. I CDH1-negative familier anbefales årlig gastroskopi med multiple biopsier og for kvinner mammografi og MR av brystene.
Gastrisk adenokarsinom og proksimal polypose i magesekken
Sist faglig oppdatert: 28.09.2021
Gastrisk adenokarsinom og proksimal polypose i magesekken eller gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS) er en meget sjelden arvelig variant av kreft i magesekken første gang beskrevet i 2012 (Rudloff, 2018). Polypose syndromet med risiko for utvikling av kreft i magesekken ble påvist i en stor familie i Australia og i to mindre familier I USA og Canada. Prevalens av dette syndromet er ukjent, men GAPPS er også påvist i Norge. Ved denne tilstanden ble det i 2016 beskrevet mutasjoner i det såkalte promotor 1B adenomatøs polyposis coli (APC) genet som hemmer genets transkripsjon. Arvegangen er autosomal dominant, men penetransen er sannsynlig ufullstendig siden noen genbærere kan ha normale endoskopifunn gjennom hele livet. GAPPS er karakterisert ved utvikling av 100 polypper eller flere i fundus- og corpusdelen av magesekken, men ikke i antrum. Karakteristiske endoskopiske funn er polypper av varierende størrelse, vanligvis mindre enn 10 mm, og kan være tilstede fra alder rundt 10 år. Tidligste mikroskopiske tegn synes å være hyperproliferative ansamlinger av spesialiserte oxyntiske celler i mucosa (de Boer, Ee, & Kumarasinghe, 2018). Pasientene med GAPPS som utvikler adenokarsinom i dysplastiske polypper er ofte unge i alder 20- og 30-årene (van der Post, Oliveira, Guilford, & Carneiro, 2019).
Diagnose
Følgende kriterier må være oppfylt for å stille diagnosen:
- Polypper avgrenset til fundus- og corpus, uten funn av polypper i duodenum, colon og rectum
- Mer enn 100 polypper hos indeks pasient, eller mer enn 30 polypper hos 1. grads slektning
- Hovedsakelig funn av funduspolypper, men noen har også funn av dysplasi eller et familiemedlem med enten dysplastiske funduspolypper eller adenokarsinom i magesekken
- Autosomal dominant arvegang
- Utelukke andre arvelige gastriske polypose syndromer og bruk av protonpumpehemmere
- Påvisning av mutasjonen i APC genet
Behandling og overvåking
Det finnes ingen konsensus på hvordan disse pasientene bør følges opp eller når man eventuelt skal tilby gastrektomi (Rudloff, 2018). Oppfølging med CT thorax/ abdomen/bekken og CT-ventriculografi, i tillegg til regelmessige gastroskopier og multiple biopsier, må balanseres mot risiko for utvikling av kreft og behov for behandling med gastrektomi. Endoskopisk oppfølging har begrensinger siden utvikling av tidlig stadium av kraft har vært oversett med påfølgende utvikling av metastaserende sykdom.
Forløpstider
Sist faglig oppdatert: 28.09.2021
Fra 1. mai 2015 ble Pakkeforløp for kreft i spiserør og magesekk innført. Da ble tidligere forløpstider erstattet av de nye tidene i Pakkeforløp for kreft i spiserør og magesekk.
Om Pakkeforløp for kreft
Sist faglig oppdatert: 28.09.2021
Pakkeforløp for kreft skal gi forutsigbarhet og trygghet for pasient og pårørende, og er et standard pasientforløp som beskriver organisering av utredning og behandling, kommunikasjon/dialog med pasient og pårørende samt ansvarsplassering og konkrete forløpstider. Pakkeforløpet starter når et helseforetak eller privat ideelt sykehus mottar en henvisning med begrunnet mistanke om kreft, eller når helseforetaket selv starter utredning med begrunnet mistanke om kreft.
Formålet med Pakkeforløp for kreft er at kreftpasienter skal oppleve et godt organisert, helhetlig og forutsigbart forløp uten unødvendig ikke-medisinsk begrunnet forsinkelse i utredning, diagnostikk, behandling og rehabilitering.
Forløpstidene i pakkeforløpet beskriver den maksimale tiden de ulike fasene i forløpet bør ta. Forløpstidene angis i kalenderdager. De enkelte fasenes forløpstid legges til slutt sammen til en samlet forløpstid, som angir tiden fra henvisning er mottatt til behandling er startet. Med utgangspunkt i pakkeforløpet skal et individuelt forløp tilrettelegges for hver enkelt pasient.
De regionale helseforetakene har det overordnede ansvaret for å sikre at pakkeforløpene med forløpstidene blir implementert og fulgt opp. Forløpstidene er normerende og er ikke en pasientrettighet. Fortsatt er det lovmessige grunnlaget pasientrettighetsloven § 2-2 (Lov om pasient- og brukerrettigheter (pasient- og brukerrettighetsloven) § 2-2 Rett til vurdering) og forskrift om prioritering av helsetjenester (Forskrift om prioritering av helsetjenester, rett til nødvendig helsehjelp fra spesialisthelsetjenesten, rett til behandling i utlandet og om klagenemnd (prioriteringsforskriften)). Av og til vil det av faglige grunner være noen pasienter som ikke kan utredes ferdig innen normert forløpstid for oppstart av første behandling. Årsaker til avvik fra de normerte forløpstidene bør dokumenters i pasientjournalen.
Forløpstider for kreft i magesekken
Sist faglig oppdatert: 28.09.2021
| Fra henvisning mottatt til første fremmøte utredende avdeling | 8 kalenderdager | |
| Fra første fremmøte i utredende avdeling til avsluttet utredning (beslutning tas) | 21 kalenderdager | |
| Fra avsluttet utredning til start behandling | Kirurgisk behandling | 14 kalenderdager |
| Fra avsluttet utredning til start behandling | Medikamentell behandling | 14 kalenderdager |
| Fra avsluttet utredning til start behandling | Strålebehandling | 14kalenderdager |
| Fra henvisning mottatt til start behandling | Kirurgisk behandling | 43 kalenderdager |
| Fra henvisning mottatt til start behandling | Medikamentell behandling | 43 kalenderdager |
| Fra henvisning mottatt til start behandling | Strålebehandling | 43 kalenderdager |
Pakkeforløp for kreft i spiserør og magesekk finnes på Helsedirektoratets nettsider og skal etter hvert også publiseres som webversjon. Se www.helsedirektoratet.no
Det er utarbeidet egne diagnoseveiledere for fastleger for inngang til pakkeforløp. Diagnoseveileder finnes på www.helsedirektoratet.no
Diagnose og utredning
Symptomer
Sist faglig oppdatert: 28.09.2021
Ved kreft i magesekken mangler spesifikke symptomer tidlig i forløpet av sykdommen. De vanligste symptomene er dyspepsi og vage øvre abdominalsmerter. Dette er en diagnostisk utfordring ettersom en i allmennpraksis vil ha et stort antall pasienter med slike symptomer. Hos pasienter med avansert sykdom foreligger ofte tretthet, appetittløshet, tidlig metthet, oppkast, vekttap og anemi. Det er typisk at smertene ikke lindres ved matinntak. Ved proksimale svulster som omfatter cardia foreligger ofte dysfagi og ved distale svulster i antrum og mot pylorus kan pasienten få ventrikkelretensjon. Om lag 40 % av pasientene har anemi og 15 % har hematemese (Townsend, Beauchamp, Evers, & Mattox, 2017). Tegn på langtkommet sykdom er ascites, palpabel tumor, og lymfeknuter i fossa supraclavicularis (Virchows glandel).
Utredning
Sist faglig oppdatert: 28.09.2021
Endoskopi
Gastroskopi med biopsier av tumorsuspekte forandringer og magesekksår gir oftest diagnosen. Ved multiple biopsier ligger diagnostisk treffsikkerhet på 98 %. Ved stiv ventrikkelvegg med redusert peristaltikk skal man mistenke diffust infiltrerende cancer. Ved gastroskopi kan stor biopsitang benyttes, evt. kan også biopsi i biopsisår utføres for å få dypere materiale. Endoskopisk ultralyd (EUS) gir informasjon om veggfortykkelse og evt. utvisking av vegglag og muliggjør også submukosale biopsier. EUS gir nyttig informasjon om stadieinndeling av tumor, og er av praktisk betydning for å skille ut kreft i tidlig stadium (S. Kelly et al., 2001).
Standardisert endoskopibeskrivelse ved mistenkt/ påvist kreft i magesekken
Ved endoskopisk undersøkelse for mistenkt eller påvist kreft i magesekken er det viktig for vurdering og planlegging av videre behandling (kurativ eller palliativ) at endoskopibeskrivelsen inneholder et minimum sett av opplysninger, formulert på en måte som gjør funnene reproduserbare og sammenlignbare (f.eks. før/etter neoadjuvant behandling), og som forhåpentligvis også kan hindre unødvendig forsinkelse i diagnostikk og behandling ved at undersøkelser må gjentas, i verste fall flere ganger.
- Tumors lokalisasjon– cardia/fundus/corpus/antrum
- Strekker tumor seg opp i øsofagus? I så fall hvor langt (i cm) i forhold til gastroøsofageal overgang
- Tumors relasjon til fremre/bakre vegg
- Tumors relasjon til major-/minorsiden
- Avstand (i cm) fra pylorus for svulster i antrum, ved gjennomvekst av pylorus vurdere innvekst i duodenum
- Tumors størrelse (i cm)
- Makroskopisk utseende (ulcererende, polyppøs, flat, linitis plastica)
- Magesekkens tøyelighet/peristaltikk
Bildediagnostisk utredning
CT-undersøkelser av thorax, abdomen og bekken bør gjøres på alle pasienter med påvist kreft i magesekken. Ventrikkelundersøkelsen bør gjøres som spesialprosedyre:
CT ventrikkel (hydro-CT eller gastrografi) utført med moderne multidetektor CT (MDCT) er en god metode for å definere tumors lokale utbredelse og relasjon til andre organer. CT er imidlertid ikke godt egnet til å skille T1/T2/T3 fra hverandre da de ulike vegglag ikke kan defineres, her har endoskopisk ultralyd høyere sensitivitet. CT er imidlertid best egnet til å påvise tumorinnvekst i nærliggende organer. Tumorutbredelse defineres best når magesekken er distendert, spesielt viktig i forhold til øsofagus, cardia og pylorus. Dette oppnås ved bruk av spasmolyticum (buscopan) intravenøst og ca. 1 liter vann / alternativt gass per os. Opptaket anbefales utført i to kontrastfaser: 30-40 s (gir best framstilling av tumor) og portvenefase ca. 70-80 s (avdekke levermetastaser). Multiplanar fremstilling med tynne snitt er nyttig for nøyaktig å definere utbredelse i magesekken og omgivelser og særlig for overgangen til øsofagus. Dediserte plan er viktig for vurdering av serosainfiltrasjon (T-staging). Intravenøs kontrast i tilstrekkelig volum og optimal flow er nødvendig for godt diagnostisk resultat (ca. 150 ml ved 3,5 ml/s) (Ba-Ssalamah et al., 2011; Kwee & Kwee, 2007; H. S. Park et al., 2010; Wei, Yu, Li, Liu, & Zheng, 2005). Undersøkelsen har høyere sensitivitet for å avdekke små tumores dersom undersøkelsen gjøres med gass i ventrikkel og man supplerer med virtuell endoskopi. Nye undersøkelser tyder på at CT volumetri av tumormasse er av verdi både for primær T-staging og terapirespons (Hallinan, 2012; S. M. Lee et al., 2009).
Flere studier viser at N-staging er unøyaktig med CT-diagnostikk. Lymfeknuter med tverrmål over 1 cm regnes for forstørrede, men forstørrede lymfeknuter er ofte reaktive og vil derfor være falske positive ved kun bruk av størrelseskriterium. Rund form er et tilleggsfunn som kan indikere malignitet. Endoskopisk ultralyd og PET har også kun tilsvarende lav/moderat sensitivitet for å avdekke lymfeknutemetastaser. I de fleste tilfeller vil en samlet vurdering fra flere modaliteter legges til grunn ved staging, likevel vil N-staging være usikker. I tvilstilfeller kan man supplere med endoskopisk ultralyd og cytologisk prøve.
Undersøkelsen bør ved verifisert malignitet utføres ved avdeling med kompetanse på diagnostikk og behandling av kreft i magesekken, basert på multidisiplinær tilnærming (Barry, Edwards, Lewis, Dhariwal, & Thomas, 2002). Ved planlagt endoskopisk behandling kan påviste suspekte lymfeknuter eventuelt undersøkes cytologisk ved målrettet endoskopisk ultralydveiledet finnålsaspirasjon.
Metodevalg ved utredning av levermetastaser avhenger av lokal kompetanse. CT lever med optimal protokoll kan påvise metastaser ned til under 5 mm. Dedikert MR lever med leverspesifikt kontrastmiddel og diffusjonssekvenser har i dag høyest sensitivitet for påvisning av små levermetastaser. Denne undersøkelsen brukes i ulik grad avhengig av tilgjengelighet og kompetanse i praksis mest som problemløser i enkelttilfeller. Den er spesielt viktig hvis leverreseksjon er aktuelt. Ultralyd lever har svært varierende sensitivitet for deteksjon av metastaser, er bruker-, pasient- og utstyrsavhengig, men kan gi et nyttig supplement for karakterisering av usikre lesjoner. UL lever med kontrast har i erfarne hender sensitivitet og spesifisitet som CT og MR. Ultralydveiledet biopsi kan eventuelt avklare usikre lesjoner påvist ved CT eller MR.
Positron emisjonstomografi (PET-CT) har vist større sensitivitet enn CT- og MR- undersøkelse i deteksjon av fjernmetastaser til lymfeknuter og skjelett ved avansert kreft (Kawanaka et al., 2016). Deteksjonsrate for levermetastaser og nøyaktighet av T-staging (Wei et al., 2005) (evidensnivå 3) er dårligere enn for CT og MR. Studier tyder på at metoden er god for påvisning av residiv av kreft i magesekken (positiv prediktiv verdi rapportert 89 %). Ut fra foreliggende litteratur har PET-CT foreløpig ingen rutinemessig plass i primærutredningen av kreft i magesekken, men kan være et supplement ved avansert kreft.
Konvensjonell dynamisk røntgenundersøkelse med peroral kontrast (gjennomlysning) har i dag ingen plass som primærdiagnostisk metode. Undersøkelsen brukes bare i enkelte tilfelle som supplement etter skopi og/eller CT for vurdering av funksjonelle forhold, f eks ved mistanke om linitis plastica, eller som kartlegging før stenting ved høygradig stenose.
Diagnostisk laparoskopi
Ved kreft i magesekken foreligger det risiko for spredning til peritonealhulen (karsinomatose), i MAGIC studien ble det funnet en uvanlig høy andel karsinomatose hos pasientene (30%) som ble randomisert direkte til kirurgi. Makroskopisk synlige peritoneale tumormasser kan forsvinne under neoadjuvant kjemoterapi som resulterer i at pasienter gjennomgår total eller subtotal gastrektomi med påfølgende raskt residiv, idet de egentlig har metastatisk sykdom på operasjonstidspunktet. Således er det indikasjon for å gjennomføre diagnostisk laparoskopi før neoadjuvant behandling på utvalgte pasienter, hvor risikoen for karsinomatose er størst. Risikoen for karsinomatose er størst hos pasienter med: svulster større enn 8 cm eller 5 cm i den gastroøsofageale overgang, Borrmann type IV svulster (linitis plastica), bulkete eller grensestore para-aortale lymfeknuter påvist ved CT, eller ved T4-svulster (Irino et al., 2018; Miki et al., 2015). I tillegg anbefales diagnostisk laparoskopi når det ved CT foreligger usikkerhet om det er karsinomatose.
Tumormarkører
Det finnes ingen diagnostiske blodprøver for kreft i magesekken.
Anbefaling
Ved mistanke om kreft i magesekken skal pasienten gis time til utredning innen åtte kalenderdager (iht. pakkeforløp).
Alle pasienter med kreft i magesekken skal undersøkes med gastroskopi og CT thorax, abdomen og bekken (evidensgrad B).
Ved påvist kreft i magesekken som vurderes for kirurgi skal pasienten utredes bildediagnostisk med tanke på stadieinndeling med CT ventrikkel, alternativt EUS (evidensgrad B).
Diagnostisk laparoskopi anbefales ved usikkerhet om karsinomatose eller ved lokalavansert tumor for pasienter som kan være aktuelle for kirurgi (evidensgrad C).
Histopatologi
Sist faglig oppdatert: 05.07.2023
95 % av maligne svulster i magesekken er adenokarsinomer, mens resten fordeles mellom lymfomer, nevroendokrine svulster (NET og NEC), gastrointestinal stromal tumor (GIST) og andre sjeldne former.
Det finnes flere klassifikasjonssystemer for inndeling av adenokarsinomer. De mest brukte er Laurèns klassifikasjon og WHO`s klassifikasjon (WHO Classification of Tumours Editorial Board, 2019). I henhold til Laurèns klassifikasjon inndeles adenokarsinomer i magesekken i intestinal, diffus (inkludert signetringcellekarsinom) og blandet type. WHO`s klassifikasjon har 5 hovedtyper og i tillegg flere sjeldne varianter. Hovedtypene er tubulær, papillær, mucinøs, lite kohesiv (inkludert signetringcellekarsinom) og blandet type.
Ved intestinal type (tubulær og papillær type, WHO) danner tumor kjertellignende eller papillære strukturerer med varierende slimproduksjon. Disse svulstene vokser typisk ekspansivt med varierende intraluminal og infiltrerende komponent. Denne svulsttypen er hyppig i endemiske områder med kreft i magesekken. Incidensen er sterkt fallende, i motsetning til diffus type, som i noen studier rapporteres å øke.
Ved diffus type (lite kohesiv type, WHO) vokser tumorcellene enkeltvis eller i små grupper med induksjon av rikelig bindevev. Ofte har cellene en stor slimvakuol i cytoplasma som forskyver kjernen til ene siden, såkalte signetringceller. Disse svulstene infiltrerer diffust i magesekkens veggog fører karakteristisk til «linitis plastica». Peritoneale metastaser er hyppig forekommende.
Sjeldne varianter utgjør ca. 5 % av svulstene. To sjeldne varianter er:
Adenokarsinom med lymfoid stroma (medullært karsinom) karakterisert av betydelig lymfocyttinfiltrasjon i tumor. Epstein-Barr virus er påvist i >80 % av svulstene. Denne varianten har relativt god prognose.
Hepatoid adenokarsinom består av hepatocytt-lignende tumorceller. AFP kan påvises i tumorcellene immunhistokjemisk og også i serum. Denne svulsten responderer dårlig på kjemoterapi og har dårlig prognose (Søreide, Greve, Gudlaugsson, & Størset, 2016).
Ved påvist adenokarsinom i en vevsprøve fra magesekken undersøkes tumor alltid med hensyn til HER-2, primært ved immunhistokjemi.
| Immunhistokjemisk | HER-2 |
|---|---|
| 0 - +1 | Negativ |
| +2 | Usikker, gjør in situ hybridisering (FISH eller SISH) |
| +3 | Positiv |
I tillegg undersøkes tumor med hensyn til MMR/MSI. Etter forespørsel fra kliniker kan tumor undersøkes med hensyn til EBV (in situ hybridisering) og PD-L1 (immunhistokjemisk).
I tillegg undersøkes tumor med hensyn til MMR/MSI. Etter forespørsel fra kliniker kan tumor undersøkes med hensyn til EBV (in situ hybridisering) og PD-L1 (immunhistokjemisk).
Ved PD-L1 testing av adenokarsinom i magesekken benyttes «Combined Positive Score» (CPS). Denne beregnes på følgende måte:
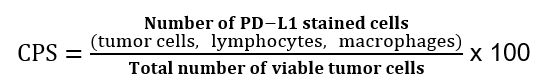
Molekylær klassifisering av gastroesofagale svulster
Sist faglig oppdatert: 28.09.2021
The Cancer Genome Atlas Project er et stort forskningsnettverk som har publisert inngående molekylær karakterisering av større tumorpaneler fra de fleste forskjellige maligniteter ved bruk av moderne metoder som nyeste generasjons dybdesekvensering, RNA sekvensering og proteomikk metoder blant annet. I 2014 publiserte nettverket en omfattende molekylær kartleggning av 295 primærsvulster fra magesekken , og inndelte disse i 4 hovedtyper; Epstein Barr virus (EBV)-positive svulster (9 % av svulstene), mikrosatellitt instabil ( MSI) –høy type (22 %), genomisk stabile svulster(20 %), og kromosomalt instabile svulster, (50 %) (Cancer Genome Atlas Research Network, 2014). De EBV positive svulstene hadde typisk PIK3CA mutasjoner, ekstrem DNA hypermetylering og amplifikasjon av JAK2, CD274 (PD-L1) og PDCDILG2 (PD-L2). Mikrosatellitt høy positive svulster hadde typisk høy mutasjonsrate i tumor-genomet. De genomisk stabile svulstene omfattet typisk den histologisk diffuse typen, og hadde oftere mutasjon i RHOA genet eller fusjonsgener som involverte gener I RHO-familien av GTPase aktiverende proteiner. Den fjerde typen, de kromosomalt instabiler svulstene var oftere aneuploide og med fokale amplifikasjoner av reseptor tyrosin kinaser.og var typisk av den histologisk intestinale typen. I 2017 publiserte det samme nettverket en tilsvarende omfattende molekylær kartlegging av 164 primærsvulster fra spiserøret, hvorav 90 var plateepitelcarcinom, resten adenocarcinom, og sammenliknet disse med svulster fra magesekken og gastroesofageal overgang, totalt over 550 karakteriserte svulster (Cancer Genome Atlas Research Network et al., 2017). De fant at de proksimale adenocarcinomene i magesekken var oftest av kromosomalt instabil type, og ingen av adenocarcinomene i spiserøret var av EBV eller MSI type. Noe lengre distalt i gastroøsofageal overgang fant man imidlertid noen av denne typen. Plateepitelcarcinom i spiserøret var en egen type, og de inndelte denne typen i 2 undergrupper. De foreslår å benytte denne molekylære karakteriseringen når man studerer pasienter i fremtidige kliniske studier av pasienter med kreft i spiserør og magesekk, for bedre stratifisering av pasientene.
Stadieinndeling
Sist faglig oppdatert: 28.09.2021
Klassifikasjon og stadieinndeling skal utføres i henhold til UICC/TNM klassifikasjon 8. utgave, 2017 (Brierley, Gospodarowicz, & Wittekind, 2017).
| TX | Primærtumor kan ikke vurderes |
| T0 | Ikke påvist primærtumor |
| Tis | Carsinoma in situ, intraepitelial tumor uten infiltrasjon i lamina propria, høygradig dysplasi |
| T1 | Tumor infiltrerer lamina propria, muscularis mucosae eller submucosa |
| T1a | Tumor infiltrerer lamina propria eller muscularis mucosae |
| T1b | Tumor infiltrerer submucosa |
| T2 | Tumor infiltrerer muscularis propria |
| T3 | Tumor infiltrerer subserosa |
| T4* | Tumor vokser gjennom serosa eller infiltrerer nabostrukturer |
| T4a | Tumor vokser gjennom serosa |
| T4b | Tumor infiltrerer nabostrukturer |
| NX | Regionale lymfeknuter kan ikke vurderes |
| N0 | Ingen regionale lymfeknuter |
| N1 | Spredning i 1-2 regionale lymfeknuter |
| N2 | Spredning i 3-6 regionale lymfeknuter |
| N3 | Spredning i 7 eller flere regionale lymfeknuter |
| N3a | Spredning i 7–15 regionale lymfeknuter |
| N3b | Spredning i 16 eller flere regionale lymfeknuter |
| M0 | Ingen fjernmetastaser |
| M1* | Fjernmetastaser |
| Merknader: T4
M1
| |
I 8. utgave av UICCs TNM klassifikasjon er det en klassifikasjon for klinisk stadium. I tillegg er patologisk stadium mer differensiert basert på endring i prognose relatert til antall patologiske lymfeknuter.
| STADIUM | T | N | M |
|---|---|---|---|
| I | T1 | N0 | M0 |
| T2 | N0 | M0 | |
| IIA | T1 | N1, N2, N3 | M0 |
| T2 | N1, N2, N3 | M0 | |
| IIB | T3 | N0 | M0 |
|
| T4a | N0 | M0 |
| III | T3 | N1, N2, N3 | M0 |
| T4 | N1, N2, N3 | M0 | |
| IVA | T4b | Any N | M0 |
| IVB | Any T | Any N | M1 |
| STADIUM | T | N | M |
|---|---|---|---|
| 0 | Tis | N0 | M0 |
| IA | T1 | N0 | M0 |
| IB | T1 | N1 | M0 |
| T2 | N0 | M0 | |
| IIA | T1 | N2 | M0 |
| T2 | N1 | M0 | |
| T3 | N0 | M0 | |
| IIB | T1 | N3a | M0 |
| T2 | N2 | M0 | |
| T3 | N1 | M0 | |
| T4a | N0 | M0 | |
| IIIA | T2 | N3a | M0 |
| T3 | N2 | M0 | |
| T4a | N1, N2 | M0 | |
| T4b | N0 | M0 | |
| IIIB | T1 | N3b | M0 |
| T2 | N3b | M0 | |
| T3 | N3a | M0 | |
| T4a | N3a | M0 | |
| T4b | N1, N2 | M0 | |
| IIIC | T3 | N3b | M0 |
| T4a | N3b | M0 | |
| T4b | N3a, N3b | M0 | |
| IV | Any T | Any N | M1 |
Inndeling av kreft i overgangen mellom spiserør og magesekk
Sist faglig oppdatert: 28.09.2021
Siewerts klassifikasjon
Siewerts klassifikasjon av cardia-nære tumores er ikke lenger relevant for prognose. Klassifikasjonen er imidlertid fortsatt nyttig til planlegging av kirurgisk behandling. Siewerts klassifikasjon baserer seg på tumorsentrums (episenter) lokalisasjon i forhold til gastroøsofageal overgang.
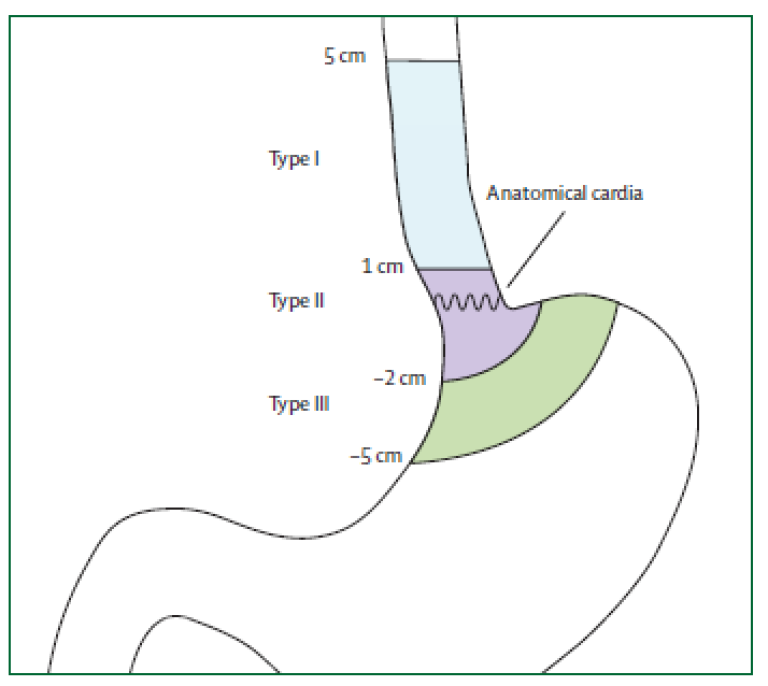
| Type I: | Sentrum av tumor proksimalt for den gastroøsofageale overgang Utgangspunkt i Barrett-slimhinne. |
| Type II: | Omfatter den gastroøsofageale overgang, både proksimalt og distalt (den egentlige cardiacancer) |
| Type III: | Lokalisert distalt for den gastroøsofageale overgang (kreft i magesekken) |
Svulster i den gastroøsofageale overgangen er nesten uten unntak adenokarsinomer. Type III cancer klassifiseres som kreft i magesekken og behandles som slik med total gastrektomi og øsofagojejunostomi. Type I cancer klassifiseres som kreft i spiserøret og behandles med øsofagusreseksjon og (vanligvis) øsofagogastrostomi. Type II cancer oppfattes nå av de fleste som spiserørskreft og behandles som dette, men noen vil fortsatt gjøre total gastrektomi og øsofagojejunostomi.
Behandling av lokalisert sykdom og Kurativ behandling
Sist faglig oppdatert: 28.09.2021
Pasienter med kreft i magesekken bør evalueres og behandling planlegges av tverrfaglig team med radiolog, kirurg, onkolog, gastroenterolog og patolog med spesiell kompetanse innenfor dette området. I tråd med dette bør evaluering og behandling av kreft i magesekken legges til sykehus hvor slik kompetanse finnes.
Behandling med kurativ intensjon er oftest multimodal. Se kapittel Onkologisk behandling for detaljer om medikamentell behandling.
Anbefaling
Pasienter med kreft i magesekken bør evalueres og behandling planlegges av tverrfaglig team med radiolog, kirurg, onkolog, gastroenterolog og helst også patolog (evidensgrad D).
Kreft i tidlig stadium
Sist faglig oppdatert: 28.09.2021
Kreft i tidlig stadium defineres som tilfeller med tumorinfiltrasjon begrenset til mukosa (T1a) eller submukosa (T1b), uavhengig av lymfeknutestatus. I Øst-Asiatiske høyinsidensland med masseundersøkelsesprogrammer utgjør denne gruppen 40-50 % av pasientene med kreft i magesekken, mens andelen i vestlige land er betydelig lavere, 6-20 % i norske reseksjonsmaterialer (Janssen, Lie, Maartmann-Moe, & Matre, 1991; Lindahl, Harbitz, & Liavåg, 1988; Wasmuth, Thorsen, Nordgård, & Gjesdahl, 1997). Sannsynligheten for lymfeknutemetastasering er < 5 % ved T1a, men øker til 11-20 % ved tumorinfiltrasjon i submukosa (T1b) (Yasuda et al., 1999).
I de japanske retningslinjene anbefales endoskopisk reseksjon når følgende kriterier er oppfylt: ikke infiltrasjon i submukosa, høyt eller middels differensiert tumor, tumor < 2 cm, ikke ulcererende tumor, ikke infiltrasjon i lymfekar (Nakajima, 2002; Tanaka et al., 2010). Det foreligger ingen randomiserte studier som sammenligner endoskopisk og kirurgisk behandling, men i observasjonsstudier har kreftspesifikk 5-års overlevelse etter endoskopisk behandling vært > 95 %, forekomsten av lokalt residiv lav og komplikasjonsraten lavere enn ved kirurgi (J. J. Kim et al., 2007; Retana, Silverstein, & Wassef, 2011). Det er også definert utvidede kriterier for endoskopisk behandling idet det har vist seg at sannsynligheten for lymfeknutemetastasering er svært liten ved infiltrasjon i submukosa når tumor er høyt eller middels differensiert og < 3 cm, eller lite differensiert, begrenset til mukosa og < 2 cm (Gotoda et al., 2000).
Forekomsten av lokalt residiv er lavere ved submukosal enn ved mukosal reseksjon, men med høyere perforasjonsrate (Oda et al., 2006). Eradikasjon av Helicobacter pylori etter endoskopisk behandling reduserer risikoen for metakron sykdom signifikant (Fukase et al., 2008). Det anbefales at pasientene følges med regelmessige endoskopiske kontroller i fem år (Kondo et al., 2001; Miyata et al., 2000).
Endoskopisk behandling av kreft i tidlig stadium stiller store krav til utredning (bl.a. endoskopisk ultralyd) og er teknisk krevende. Med den lave forekomsten vi har i Norge bør denne behandlingen samles på få steder. Endoskopisk behandling er tilstrekkelig hvis følgende kriterier er oppfylt:
- Tumores som er begrenset til mukosa (cT1a) eller øvre del av submukosa (cT1b)
- Lesjoner < 2 cm
- Høyt eller middels differensiert tumor
- Ikke ulcererende tumor
- Ikke påvist patologisk forstørrede lymfeknuter
Hos høyrisikopasienter kan endoskopisk behandling også vurderes ved:
- Tumores som infiltrerer dypere i submukosa, som er høyt eller middels differensiert og som er < 3 cm
- Tumores som er lite differensiert, begrenset til mukosa (cT1a) og er < 2 cm
Hvis histologisk undersøkelse av reseksjonspreparatet viser at kriteriene ikke er oppfylt, må risikoen ved en formell ventrikkelreseksjon veies opp mot risikoen for residiv.
Anbefaling
- Pasienter med kreft i tidlig stadium bør vurderes for endoskopisk behandling (evidensgrad D).
- Endoskopisk behandling av kreft i tidlig stadium bør sentraliseres til få sykehus (evidensgrad D).
- Det anbefales at pasientene følges med regelmessige endoskopiske kontroller i fem år (evidensgrad D).
Kirurgisk behandling
Sist faglig oppdatert: 28.09.2021
Operatør bør ha gastroskopert pasienten, slik at en kan danne seg et så nøyaktig bilde som mulig av svulstens lokalisasjon og utbredelse.
Innvekst av svulsten til bakre bukvegg, metastaser til lever (Kakeji, Morita, & Maehara, 2010), lunge og peritoneum og/eller påvisning av større mengder ascites er oftest kriterier for irresektabilitet.
Valg av operasjonsmetode
Ved lokalisasjon av svulsten i proksimale del av magesekken (Siewert type III) gjøres total gastrectomi og øsofagojejunostomi. Det samme (evt. med torakotomi for tilstrekkelig proksimal reseksjonskant) kan også være aktuelt ved Siewert type II selv om de fleste nå vil behandle denne som spiserørskreft med øsofagusreseksjon/proksimal ventrikkelreseksjon og øsofagogastrostomi. Det skal utføres frysesnitt av reseksjonsrand mot spiserøret, ettersom det er vist at så mange som 13 % av opererte pasienter ikke har fri øvre reseksjonskant mot spiserøret ved kun makroskopisk vurdering av resektat (Hallissey, Jewkes, Dunn, Ward, & Fielding, 1993). Resultater av rekonstruksjon med anleggelse av reservoar etter gastrektomi har nylig vært rapportert i en metaanalyse basert på 25 sammenlignende studier av 1651 pasienter (Syn, Wee, Shabbir, Kim, & So, 2019). Sytten av undersøkelsene var randomiserte kontrollerte studier. Halvparten av pasientene fikk et reservoir (n=873), vanligvis jejunalt J-reservoir, og resultatene ble sammenlignet med pasientene uten reservoar etter gastrektomi. Ved postoperativ observasjonstid fra 1 til 2 år hadde pasientene med reservoar mindre forekomst av problemer med matinntak (14% vs. 44%), dumping syndrom (3% mot 24%), halsbrann (3% vs. 12%) og noe bedre livskvalitet. Parametere som hemoglobin, serumjern var uforandret mellom gruppene, mens forskjellen i BMI og albumin var marginale og uten klinisk betydning. I en eldre langtids-studie etter 5 år forelå ingen dokumentasjon for nytte av reservoar (Liedman, Bosaeus, Hugosson, & Lundell, 1998). Det forelå ingen forskjell i morbiditet og mortalitet mellom gruppene, men operasjonstiden var lengre hos reservoaropererte. Bruk av reservoir etter gastrektomi benyttes sjelden. Operasjonen øker kompleksiteten av inngrepet og anbefales kun ved spesielle indikasjoner og utført av erfaren operatør.
Ved kreft i corpus eller antrum utføres ved intestinal type subtotal gastrektomi. Ved diffus type må en enten utføre total gastrektomi eller sikre fri reseksjonskant med frysesnitt. Det er vist at proksimal reseksjonsmargin på 5 cm fra tumors palpable avgrensning er tilstrekkelig både for intestinal og diffus tumortype. Imidlertid krever svulster som har penetrert serosa (T4a) en større reseksjonsmargin, og en margin på 6 cm fra tumors proksimale avgrensning for begge typer er anbefalt (Bozzetti et al., 1982)
Ved kreft i distal del utføres subtotal gastrectomi uten splenektomi. Splenectomi øker postoperativ morbiditet og mortalitet (Viste, Haùgstvedt, Eide, & Soreide, 1988).
Ved innvekst i naboorganer anbefales reseksjon en bloc når operasjonen kan utføres kurativt.
Radikal laparoskopisk ventrikkelreseksjon og gastrektomi har vunnet stor utbredelse. Omlegging fra åpen til laparoskopisk tilgang er fundert på en antagelse om lavere sykelighet og like gode langtidsresultater. Enn så lenge er de fleste publikasjoner retrospektive sammenlignende studier og det er få og små randomiserte studier. Noen større sammenstillinger refereres her. I en randomisert studie av 296 pasienter med kreft i magesekken (alle lokalisasjoner) gikk 128 til laparoskopisk reseksjon (LR) og 142 til åpen reseksjon (ÅR) (Cui et al., 2015). Antall lymfeknuter som ble høstet ved LR (29,3±11,8) var temmelig likt antallet ved ÅR (39,1 ±11,4) (p=0,574). Det var noe mindre blødning ved ved LR enn ÅR (258±80 min vs 194 ± 49), p<0,001). Tid til første mobilisering og flatus var statistisk kortere ved LR enn ÅR. Liggetiden var lang i begge grupper og henholdsvis 14 dager i LR og 18 dager ved ÅR (p=0,005).
En studie med randomisert inklusjon av 1056 pasienter vurderte sykelighet og dødelighet ved (LR) og åpen (ÅR) D2 reseksjon av fremskredet distal ventrikkelcancer (Hu et al., 2016). Primærendepunkt og utvalgstørrelse var basert på 3 års sykdomsfri overlevelse. Operasjonstiden var signifikant lengre (217 min ± 60 vs 186 min ± 53) ved LR og blødning mindre (105 ml ± 88 vs 117 ± 84) ved LR. Intraoperative komplikasjoner oppstod hos 4,8 % etter LR og 3,5 % etter ÅR. Postoperative komplikasjoner oppstod blant 15,2 % ved LR og 12,9 % ved ÅR, og dødelighet var henholdsvis 0,4 % og 0. Det var ingen statistisk forskjell i alvorlighet av komplikasjoner etter Clavien Dindo’s klassifikasjon.
En metaanalyse i regi av Cochrane vurderte laparoskopisk versus åpen gastrektomi ved kreft i magesekken (Best, Mughal, & Gurusamy, 2016). 2528 pasienter hvorav 1288 LR og 1240 ÅR. Konklusjonen var at evidensgrunnlaget var lavt eller svært lavt. Død innen 30 dager var 6/1000 ved LR og 3/1000 ved ÅR, men de kunne ikke konkludert med noen forskjell. Data på alvorlige komplikasjoner innen 3 mnd (LR: 36/1000 vs 60/1000 ÅR) eller alle komplikasjoner (LR 161/1000 versus ÅR 253/1000), behov for transfusjon og liggetid i sykehus var alle upresise. De kunne derfor ikke konkludere med fordeler eller øket risiko ved laparoskopisk- eller åpen reseksjon.
Langtidsoverlevelse finnes så langt kun fra retrospektivt sammenlignende studier. I en studie av 1874 pasienter med tidlig eller avansert ventrikkelcancer (alle lokalisasjoner) hvor 816 ble behandlet med ÅR og 1058 med LR ble det ikke funnet forskjeller i sykdomsfri overlevelse (J. H. Lee et al., 2014).
Splenektomi
Flere randomiserte kontrollerte studier har vurdert behovet for splenektomi ved operasjon for kreft i magesekken. I en studie av 505 pasienter med proksimalt beliggende svulst uten tumorinfiltrasjon av curvatura major (T2-4/N0-2/M0) var splenektomi forbundet med høyere sykelighet og blodtap uten at 5-års overlevelse var påvirket, henholdsvis 75,1 % og 76,4 % i splenektomi og miltbevarende gruppe (Sano et al., 2017). Økt sykelighet og manglende effekt på overlevelse er også funnet i en annen RCT av 207 pasienter (W. Yu, Choi, & Chung, 2006). 5- års overlevelse var her 54,8 % etter splenektomi og 48,8 % etter miltbevarende kirurgi. I dette materialet forelå ingen langtidsoverlevere blant pasienter med metastaseinfiltrerte lymfeknuter i milthilus (stasjon 10), selv om det ble utført fjerning av lymfeknuter ved stasjon 10 med eller uten splenektomi.
Splenektomi kan vurderes ved proksimal tumor og lokalisasjon mot magesekkens majorside og bør utføres ved mistanke om metastatiske lymfeknuter i milthilus eller direkte tumorinnvekst i milten (Allum et al., 2011).
Omentektomi
Det foreligger i dag ingen evidens for at omentektomi ved total eller subtotal gastrektomi gir noen overlevelsesgevinst. Japanske retningslinjer anbefaler at man deler det gastrokoliske ligament minimum tre cm fra gastroepiploica-karene, for å få med lymfeknutene svarende til stasjon 4. Det pågår en randomisert studie i Japan (Sato et al., 2020) hvor pasienter med T3 og T4 svulster randomiseres til omentsparende eller ikke-omentsparende gastrektomi. Bursektomi har tidligere ikke vist noen overlevelses gevinst (Kurokawa et al., 2018). For øvrig er det publisert to japanske retrospektive studier og en koreansk propensity matched studie (D. J. Kim, Lee, & Kim, 2014), som ikke viser overlevelsesgevinst ved omentektomi.
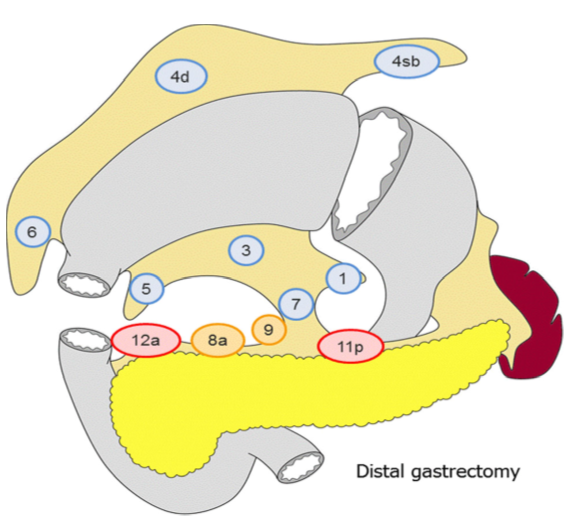
Lymfeknutedisseksjon
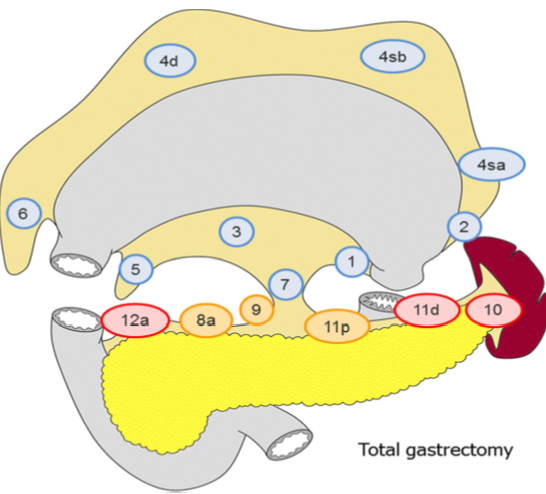
D2 lymfadenektomi omfatter etter Japanske retningslinjer (Japanese Gastric Cancer Association, 2017) stasjon 1, 3, 4sb, 4d, 5, 6, 7, 8a, 9, 11p og 12a ved subtotal gastrektomi og stasjon
1-7, 8a, 9, 11p, 11d og 12a ved total gastrektomi. I Europa og Vesten har det imidlertid ikke vært praksis å ta med miltnære lymfeknuter (stasjon 10), med mindre det foreligger indikasjon for splenektomi på grunn av innvekst av tumor.
I randomiserte studier har man påvist økt komplikasjonsfrekvens etter utvidet lymfeknutedisseksjon (D2 fremfor D1 reseksjon). Det skal bemerkes at det i disse studiene ved D2 som standard ble utført milt- og pankreasreseksjon. Det ble ikke funnet forskjell i 5-års overlevelse mellom de to metodene. Imidlertid har man nå fra den nederlandske studien publisert resultater etter 15 års observasjonstid. Man finner da signifikant reduksjon i lokalt og regionalt residiv samt bedret kreftrelatert overlevelse (29% versus 21%) ved D2-reseksjon (Songun, Putter, Kranenbarg, Sasako, & van de Velde, 2010). Det anbefales derfor at pasienter som får utført kurativ operasjon for kreft i magesekken opereres med D2 lymfeknutedisseksjon (minus stasjon 10) uten standard fjerning av milt eller distal pankreasreseksjon.
Antall lymfeknuter som ekstirperes/undersøkes er en god kvalitetsparameter både for kirurgien og patologien. Dette gir en pålitelig stadieinndeling som kan være nyttig for senere forskning, og det er en sammenheng mellom antall undersøkte lymfeknuter og langtidsoverlevelse (Ichikura et al., 2003). Det bør derfor fjernes og undersøkes minst 16 lymfeknuter.
Figur hentet fra Japanese gastric cancer treatment guidelines 2014 (Japanese Gastric Cancer Association, 2017)
Anbefaling
- Siewert type II cardiacancer behandles enten som spiserørskreft alternativt med total gastrektomi og distal øsofagusreseksjon
- Siewert type III behandles med total gastrektomi (evidensgrad D).
- Splenektomi kan vurderes ved proksimal tumor og lokalisasjon mot magesekkens majorside og bør utføres ved mistanke om metastatiske lymfeknuter i milthilus eller direkte tumorinnvekst i milten (evidensgrad B).
- Ved intestinal type kreft i corpus og antrum kan det utføres subtotal gastrektomi (evidensgrad A).
- Ved diffus type må det enten utføres total gastrektomi eller subtotal gastrektomi, forutsatt fri reseksjonskant ved frysesnitt (evidensgrad A).
- Ved kreft i distale del utføres reseksjon uten splenektomi (evidensgrad A).
- Subtotal gastrektomi bør foretas, hvor man kan oppnå fri proksimal reseksjonsrand på minst 6 cm (evidensnivå A).
- Ved total gastrektomi skal det alltid utføres frysesnitt av reseksjonsranden (evidensgrad D).
- Resultatene ved laparoskopisk kirurgi synes så langt likeverdige med åpen tilgang (evidensgrad B).
- Ved kurativ kirurgi utføres modifisert D2 lymfeknutedisseksjon (evidensgrad A).
Besvarelse av ventrikkelresektater
Opplysning på remissen som følger preparatet:
Neoadjuvant behandling: ja/nei
Operasjonstype
Håndtering av preparatet før fiksering:
Oppklipping av preparatet langs curvatura major og oppspenning på korkplate
Besvarelse fra patologen:
Tumors histologiske type
Differensieringsgrad
Tumors største diameter på overflaten
Infiltrasjonsdybde
Avstand til proksimale og distale reseksjonsrand
Avstand til sirkumferent reseksjonsrand ved curvatura major og minor
Tumorinfiltrasjon i kar/perinevralt
Antall lymfeknuter med metastaser/totalt antall lymfeknuter
Evt. neoadjuvant behandlingseffekt
SNOMED-kode
TNM-klassifisering
Konferer forøvrig
Veileder i biopsibesvarelse av maligne svulster. 3. utg. Oslo: Den norske patologforening; 2016.
Postoperativt regime
Pasienten kan innta væske peroralt umiddelbart etter total gastrektomi, eller så snart det ikke er tegn til ventrikkelretensjon ved subtotal gastrectomi. Anastomosekontroll med vannløselig kontrast utføres ved klinisk mistanke om lekkasje (Mortensen et al., 2014).
Komplikasjoner
Av spesifikke kirurgiske komplikasjoner forekommer intraabdominale infeksjoner hos 5-10 % og sårinfeksjoner hos 3–5 % av pasientene. Anastomoselekkasje etter total gastrectomi finnes hos under 5 % av pasientene (Hallissey et al., 1993) og 90-dagers mortalitet bør være under 5 %.
Resultater
Standardisert preoperativ utredning, særlig med bruk av dedikert CT-undersøkelse, evt. supplert med eksplorativ laparoskopi, bør i høy grad identifisere pasienter som egner seg for kurativ kirurgi, palliative reseksjoner, stenting eller ingen intervensjon. Eksplorative laparotomier bør ikke forekomme hyppigere enn i 5 % av tilfellene. Fem års total overlevelse hos kurativt opererte er 35-50 % (Bringeland et al., 2013). Det er i flere studier dokumentert at resultatene er bedre i høyvolumsentra enn i enheter som gjør et lite antall inngrep for kreft i magesekken (Halm, Lee, & Chassin, 2002; Killeen, O'Sullivan, Coffey, Kirwan, & Redmond, 2005).
Perioperativ onkologisk behandling
Sist faglig oppdatert: 28.09.2021
Kreft i magesekken er ved diagnosetidspunktet ofte avansert. Selv ved radikal kirurgi vil en stor andel få tilbakefall, og multimodal kombinasjonsbehandling er i dag standard for stadium ≥ IB sykdom. Det foreligger ingen internasjonal konsensus om hvilken framgangsmåte som er å foretrekke, og det er ulike tradisjoner i forskjellige deler av verden.
I Asia er adjuvant (postoperativ) kjemoterapi standard basert bl.a. på den japanske ACTS-GC studien (Sasako et al., 2011). Adjuvant kombinert radio-kjemoterapi var tidligere standard i USA basert på INT0116 studien (Macdonald et al., 2001). I Europa ble perioperativ kjemoterapi tatt i bruk etter publisering av MAGIC studien i 2006 (Cunningham et al., 2006).
Perioperativ kjemoterapi ved resektabel kreft i magesekken
MAGIC studien påviste en bedring i 5-års overlevelse fra 23 % til 36 % for pasienter med resektabel stadium II og III gastro-øsofageal kreft som ble behandlet med 6 kurer (3 pre- og 3 postoperativt) perioperativ ECF (epirubicin, cisplatin og kontinuerlig infusjon med 5-FU) sammenlignet med kirurgi alene. Studien inkluderte totalt 503 pasienter med adenokarsinom i distale spiserør (11 %), cardia (15 %) eller magesekk (74 %). Det var imidlertid kun 55 % som startet den postoperative behandlingen og 42 % som fullførte alle 6 kurene (Cunningham et al., 2006).
Den franske FNLCC/FFCD studien viste en tilsvarende fordel med perioperativ kjemoterapi der 224 pasienter med resektabelt stadium II eller høyere adenokarsinom i distale spiserør (11 %), cardia (64 %) eller magesekk (25 %) ble randomisert til 2 – 3 kurer med preoperativ og 3 – 4 kurer postoperativ kjemoterapi (cisplatin og 5-døgns infusor 5-FU hver 4. uke) eller kirurgi alene. Mant fant at 5-års overlevelse økte fra 24 % til 38 %. Bare halvparten av de som fikk minst 1 preoperativ kur startet postoperativ kjemoterapi (Ychou et al., 2011).
I en metaanalyse fra 2013 fant man at perioperativ kjemoterapi for resektabel adenokarsinom i spiserør, cardia og magesekk økte overlevelse sammenlignet med kirurgi alene. (HR 0.81; 95 % CI 0.73 - 0.89). Det var en trend for større overlevelsesgevinst for svulster i cardia sammenlignet med spiserør og magesekk (Ronellenfitsch et al., 2013).
Med bakgrunn i MAGIC studien ble perioperativ kjemoterapi innført som rutine i Norge ved resektabel kreft i magesekken stadium II – III i 2007.
Basert på REAL studien som viste at capecitabin er minst like effektivt som infusor-behandling med 5-FU og oxaliplatin er minst like effektivt som cisplatin i metastatisk setting, kan capecitabin erstatte infusor 5-FU og oxaliplatin er ansett som likeverdig med cisplatin (Sumpter et al., 2005).
I en retrospektiv gjennomgang av pasienter behandlet for kreft i magesekken i helseregion Midt-Norge har man på «intention-to-treat» basis sammenlignet langtidsoverlevelse før og etter innføring av perioperativ kjemoterapi i 2007. 91 pasienter ble identifisert i perioden 2001-2007, og disse ble planlagt for kun kirurgi, mens 100 pasienter ble identifisert i perioden 2007-2011, og disse ble planlagt for perioperativ kjemoterapi med ECX/EOX regime med 3 + 3 kurer før/etter kirurgi. Man observerte en ikke ubetydelig toksisitet av kjemoterapien. Kun 44 % fullførte alle kurene. Det ble ikke observert noen negativ innvirkning av perioperativ kjemoterapi på kirurgisk morbiditet eller mortalitet. Selv om man observerte en objektiv tumorrespons på kjemoterapi, fant man ingen bedring i langtidsoverlevelse etter innføring av perioperativ kjemoterapi. 5-års overlevelse var rundt 41 % i begge periodene (Bringeland, Wasmuth, Fougner, Mjones, & Gronbech, 2014).
Den tyske FLOT4-AIO studien randomiserte over 700 pasienter med adenokarsinom i magesekk eller gastroøsofageal overgang med klinisk stadium cT2 eller høyere eller N+ og M0 til perioperativ kjemoterapi med 3 neoadjuvante og 3 adjuvante ECX/ECF kurer i standardarmen mot 4 kurer neoadjuvant og 4 kurer adjuvant med FLOT i den eksperimentelle armen. Fase II data ble publisert i 2016 (Al-Batran et al., 2016) og viste at histologisk komplett respons i operasjonspreparatet var 16 % for pasienter i FLOT-armen versus 6 % i standardarmen. Fase III delen av studien ble publisert 2019, og viser overlevelsesgevinst for pasienter behandlet med FLOT4 regimet perioperativt sammenliknet med ECX/ECF, med median overlevelse 50 mnd, sammenliknet med 35 mnd for ECX/ECF. Estimert 5-års overlevelse er 45 % i FLOT gruppen og 36 % i ECX/ECF gruppen. Antall pasienter med alvorlig toksisitet var likt i begge armer (27 % vs 27 %). Hospitalisering for toksisitet forekom hos 26 % i ECF/ECX arm vs 25 % i FLOT-arm (Al-Batran et al., 2019).
FLOT4-AIO har medført at perioperativ kjemoterapi med ECX eller EOX regimet er erstattet med FLOT-regimet. Dublett med et platinum og 5-FU (f. eks. FOLFOX eller CiFu) kan vurderes om man forventer at trippelregimet blir for toksisk.
Det er ikke vist at anti-EGFR eller antiangiogenetiske medikamenter har effekt i neoadjuvant/adjuvant situasjon. Trastuzumab og evt. Pertuzumab i perioperativ behandling av HER-2 positiv lkreft i magesekken studeres i den pågående INNOVATION studien (Neoadjuvant Study Using Trastuzumab or Trastuzumab With Pertuzumab in Gastric or Gastroesophageal Junction Adenocarcinoma (INNOVATION), 2015-2024). En annen fase II/III studie, PETRARCA, studerte perioperativ behandling av HER-2 positiv kreft i magesekken med tillegg av trastuzumab og pertuzumab konkomitant med FLOT perioperativt samt 9 sykler adjuvant med antistoffene hver 3. uke i den eksperimentelle armen. Studien var forskerinitiert (tyske AIO), ble lukket tidlig etter kun 81 pasienter randomisert, men nådde likevel sitt primære endepunkt for fase II delen, med signifikant høyere andel pasienter med patologisk komplett respons med tillegg av trastuzumab/pertuzumab sammenliknet med FLOT regimet alene (35% vs 12%, p=0,02). HR for sykdomsfri overlevelse (DFS var 0,58, 95% CI (0,278-1,139) i favør av kombinasjonen FLOT/trastuzumab/pertuzumab, men uten tilstrekkelig statistisk styrke for å kunne evaluere fase III delen av studien (Honório et al., 2020). Betydningen av anti-HER2 rettet behandling i neo-/adjuvant situasjon er således foreløpig uavklart, og kan ikke anbefales på generelt grunnlag.
Anbefaling
Det anbefales at pasienten opereres 4-6 uker etter avsluttet neoadjuvant kjemoterapi.
Adjuvant gis tilsvarende som neoadjuvant kjemoterapi. Den postoperative cytostatika behandlingen bør startes innen 6-8 uker etter operasjon, senest innen 12 uker.
Adjuvant medikamentell behandling
Meta-analyser har vist at adjuvant cytostatikabehandling etter R0 reseksjon for kreft i magesekken kan gi forlenget overlevelse (Earle & Maroun, 1998).
I en meta-analyse fant man størst reduksjon i dødsrisiko i fem studier utført i Asia (relativ risiko (RR) 0.74, 95 % CI 0.64-0.85), mens effekten i 14 studier utført utenfor Asia var mindre (RR 0.90, 95 % CI 0.85-0.96) (Liu, Wang, Chen, & Sun, 2008).
I en annen meta-analyse, hvor data fra 17 randomiserte studier med 3838 pasienter var inkludert, fant man at pasienter som fikk adjuvant cytostatikabehandling hadde 18 % redusert risiko for tilbakefall, utvikling av en ny primærtumor eller død innen 5 år (Paoletti et al., 2010). Det var 6 % økt overlevelse i favør av adjuvant cytostatikabehandling. De fleste av pasientene fikk kombinasjonsregimer som inneholdt 5-FU.
En Cochrane review fra 2013 evaluerte data fra 34 randomiserte studier som sammenlignet adjuvant systemisk kjemoterapi versus kirurgi alene, utført i både asiatiske og vestlige populasjoner. Risiko for død hos pasienter som fikk adjuvant kjemoterapi ble redusert med 15 prosent (HR for død 0,85, 95% CI 0,80 - 0,90) (Diaz-Nieto, Orti-Rodriguez, & Winslet, 2013).
I den japanske ACTS-GC studien ble 1059 pasienter med kreft i magesekken stadium II-III - etter gastrektomi med D2 disseksjon - randomisert til enten 12 måneders behandling med S1 (et peroralt fluoropyrimidin preparat) eller observasjon alene. Man fant at fem års overlevelse økte fra 61 % i gruppen for kirurgi alene til 71.7 % i gruppen som fikk adjuvant kjemoterapi (Sakuramoto et al., 2007; Sasako et al., 2011). Effekten av S1 i adjuvant setting er ikke dokumentert i vestlige studier.
I CLASSIC studien ble 1035 pasienter fra Sør-Korea, Kina og Taiwan med kreft i magesekken stadium II-IIIB - etter D2 gastrektomi - randomisert til 6 måneders adjuvant behandling med capecitabin i kombinasjon med oxaliplatin eller observasjon alene (Bang et al., 2012). Det var en statistisk signifikant forbedret fem-års overlevelse med kjemoterapi på 78 % versus 69 % med kirurgi alene (Noh et al., 2014).
Bare 67 % av pasientene som fikk kjemoterapi fikk alle åtte sykluser som planlagt, og bivirkninger (oftest nøytropeni, kvalme, oppkast, trombocytopeni, og anoreksi) førte til dosejusteringer hos 90 % av pasientene.
Ovenstående data tyder på at kjemoterapi med et fluoropyrimidin-holdig regime gir en overlevelsesgevinst etter R0 ventrikkelreseksjon ved kreft i magesekken stadium II - III.. Effekt av adjuvant kjemoterapi etter gastrektomi med D2 disseksjon er ikke bekreftet i vesten. Historisk har man observert en større gevinst med adjuvant kjemoterapi i asiatiske studier.
For enkelte pasienter i god allmenntilstand som er operert uten neoadjuvant kjemoterapi, kan man vurdere adjuvant kjemoterapi ved kreft i magesekken stadium Ib-III.. Hvilket cytostatika regime man bør velge er uklart. Alternative regimer er CAPOX/FLOX/FOLFOX i 6 mnd. Ved dårlig toleranse for oxaliplatin kan man vurdere å gi fluoropyrimidin monoterapi.
Neoadjuvant radiokjemoterapi ved kreft i gastroøsofageale overgang kan i noen tilfeller være aktuelt, gitt i henhold til CROSS studien der 58% av pasientene hadde kreft i distale spiserør (Siewert I) og 24% i gastroøsofageale overgang (tumor involverte cardia ventriculi -Siewert II/III) (Shapiro et al., 2015; van Hagen et al., 2012). I CHECKMATE577 studien studerte man nytten av adjuvant nivolumab etter operasjon for kreft i spiserøret eller kreft i gastroøsofageale overgang som hadde fått neoadjuvant radiokjemoterapi. 794 pasienter som ikke hadde patologisk komplett respons etter CROSS neoadjuvuant radiokjemoterapi, ble randomisert 2:1 til enten nivolumab adjuvant i ett år, eller placebo (R. J. Kelly et al., 2021). Gruppen med nivolumab hadde signifikant forlenget sykdomsfri overlevelse (DFS), median 22,4 mnd vs 11mnd for placebo-gruppen (p=0,0003), med en 31% redusert risiko for progresjon eller død (R. J. Kelly et al., 2020). Endelig publikasjon avventes.
Adjuvant stråle- og cytostatikabehandling
Den amerikanske INT0116 studien fra 2001 viste at adjuvant behandling med 5-FU/ kalsiumfolinat og fraksjonert strålebehandling (45 Gy gitt med 25 fraksjoner) ga 9 måneders (36 vs 27 mnd.) forlenget median overlevelse sammenlignet med kirurgi alene. 559 pasienter med ≥ T3 og/eller N+ adenokarsinom i cardia eller magesekk ble etter R0 reseksjon randomisert til radiokjemoterapi eller observasjon (Macdonald et al., 2001). Etter 10 års oppfølging observerte man fortsatt en signifikant overlevelsesgevinst med fem-års overlevelse på 43 % versus 28 % til fordel for adjuvant radio-kjemoterapi (Smalley et al., 2012).
På bakgrunn av denne studien ble postoperativ radiokjemoterapi innført som rutine i USA, men denne behandlingen fikk aldri særlig innpass i Europa. Det har vært reist bekymring i forhold til potensielle senskader etter strålebehandling samt kvaliteten på kirurgien i studien. Studien har blitt kritisert for inadekvat kirurgi ved at over 50 % av pasientene hadde mindre enn D1 lymfeknute disseksjon, og at postoperativ radio-kjemoterapi muligvis kun kompenserer for suboptimal kirurgi (Smyth et al., 2016).
I ARTIST studien fra Sør-Korea sammenlignet man adjuvant kjemoterapi mot adjuvant kombinert kjemoradioterapi. 458 pasienter etter radikal reseksjon med D2 lymfeknutedisseksjon for kreft i magesekkenr ble randomisert. Kjemoterapi armen bestod av 6 tre-ukers sykler med cisplatin - capecitabin (XP), mens kjemo-radioterapi armen besto av 2 sykler XP etterfulgt av 45 Gy med konkomitant capecitabin gitt over 5 uker og tilslutt ytterligere 2 sykler XP (Dikken et al., 2011). Etter 7 års oppfølging var det fortsatt ingen forskjell i DFS og 5-års overlevelse var rundt 74 % i begge gruppene. Subgruppeanalyser viste at kjemoradioterapi betydelig forbedret DFS hos pasienter med lymfeknute-positiv sykdom og hos pasienter med intestinal-type (S. H. Park et al., 2015). I ARTIST II studie ble kun pasienter med lymfeknutepositiv sykdom inkludert. Adjuvant kjemoradioterapi ble sammenlignet med to ulike adjuvante kjemoterapiregimer. Det var ingen forskjell i DFS mellom SOX (S-1 + oxaliplatin) i 6 mndr. og SOXRT (SOX og kjemoradioterapi til 45 Gy). DFS var ved 3 år hhv. 78 % og 73 %. Disse 2 behandlinsregimene ga imidlertid signifikant forlenget DFS sammenlignet med S-1 monoterapi (DFS 65 %) (S. H. Park et al., 2019).
I tillegg til ARTIST studiene over er det gjort flere (hovedsakelig asiatiske) randomiserte studier som sammenligner effekten av adjuvant kjemoterapi mot adjuvant radiokjemoterapi (Bamias et al., 2010; T. H. Kim et al., 2012; Kwon et al., 2010; C. Yu, Yu, Zhu, Song, & Li, 2012; Zhu et al., 2012). Med unntak av en mindre studie (C. Yu et al., 2012) er det ikke så langt vist noen overlevelsesgevinst ved å legge til strålebehandling til adjuvant kjemoterapi.
I den nederlandske CRITICS-studien sammenlignes perioperativ kjemoterapi etter MAGIC-studiens opplegg mot samme neoadjuvante kjemoterapi med adjuvant radioterapi med konkomitant cisplatin og capecitabin ved resektabel kreft i magesekken stadium Ib – III.. Kirurgien er standardisert og innebærer modifisert D2 lymfeknutedisseksjon (Dikken et al., 2011). 788 pasienter ble randomisert. Ved median oppfølging på 61,4 måneder, var median overlevelse 43 mnd i kjemoterapi-arm og 37 mnd i kjemoradioterapi-arm (HR 1,01;p=0,90 (Cats et al., 2018).
Tilleggsgevinst av postoperativ radiokjemoterapi hos pasienter som har fått neoadjuvant kjemoterapi er således ikke påvist.
I den pågående TOPGEAR studien sammenlignes perioperativ kjemoterapi etter MAGIC-studieopplegg mot neoadjuvant kjemoterapi + radiokjemoterapi og adjuvant kjemoterapi ved resektabel kreft i magesekk og cardia stadium Ib – III (unntatt T2N0). (Leong et al., 2015).
Da nytte av adjuvant radiokjemoterapi etter moderne kirurgi med adekvat lymfeknutedisseksjon ikke er bekreftet i vestlige studier, anbefales ikke dette innført som rutinebehandling.
Ved histologisk ufrie reseksjonsrender kan også postoperativ kombinert radio-kjemoterapi vurderes. Denne bør startes så snart som mulig etter kirurgi, helst innen 6-8 uker og senest innen 12 uker. En retrospektiv sammenligning av den nederlandske D1/D2 studien har antydet signifikante forbedringer i total overlevelse og forekomst av lokalt residiv med bruk av radiokjemoterapi etter R1 reseksjon. Dette har blitt bekreftet av andre retrospektive serier (Dikken et al., 2010; Stiekema et al., 2014). Det finnes imidlertid ingen sikker dokumentasjon for at radiokjemoterapi i denne situasjonen påvirker overlevelsen. En må også ta hensyn til at strålebehandling i denne regionen medfører toksisitet fra tarm, lever og nyrer.
Ved postoperativ radiokjemoterapi bør strålebehandlingen fortrinnsvis gis med konkomitant 5-FU basert kjemoterapi til en total dose på 45 Gy i 25 fraksjoner og med 5 fraksjoner per uke med intensitet-modulert stråleterapiteknikker (Trip et al., 2014). Målvolumet bør omfatte operasjonsområdet med tumorseng og ev. ventrikkelrest, anastomoseområdet og nærmeste lymfedrenasjeområde (Dikken et al., 2011; C. Yu et al., 2012). Konkomitant gis 5-FU og kalsiumfolinat eller capecitabine.
Perioperativ eller adjuvant onkologisk behandling for alle?
Det finnes pasientgrupper der effekten av perioperativ eller adjuvant onkologisk behandling er mindre eller usikker.
Mikrosatellitt instabile svulster (MSI)
I en retrospektiv analyse fra Magic studien fant man at svulster med dMMR (deficient mismatch repair) og/eller mikrosatellitt instabilitet (MSI) var assosiert med positiv prognostisk effekt hos pasienter med resektabel kreft i magesekken og gastroøsofageal overgang som ble behandlet med kirurgi alene og negativ prognostisk effekt hos pasienter som fikk perioperativ cellegiftbehandling. Antallet pasienter med MSI og/eller dMMR i Magic studien var relativ lavt (Smyth et al., 2017). I Classic studien hadde ikke MSI pasienter bedret sykdomsfri overlevelse etter adjuvant behandling (Choi et al., 2019), mens mikrosatellitt stabile (MSS) pasienter som samtidig var PDL1 negative hadde signifikant bedret sykdomsfri overlevelse etter adjuvant behandling. Overlevelsesdata fra FLOT studien for subgruppene av pasienter som hadde MSI fenotype sammenlignet med mikrosatellitt stabil (MSS) fenotype foreligger ikke enda. Vi kan således ikke sikkert si om perioperativ cellegiftbehandling med FLOT bør gis til pasienter som har MSI, eller om disse pasientene heller bør anbefales kirurgi alene. Inntil ytterligere data foreligger, anbefales perioperativ kjemoterapi for pasienter med både MSI og MSS svulster.
Diffus type kreft i magesekken
Fase II data i FLOT4-AIO studien viste patologisk komplett respons hos 16 % av pasientene som mottok FLOT og 6 % av pasientene som mottok ECF/ECX. Det var stor forskjell i histopatologisk regresjon mellom diffus og intestinal type tumor. Pasienter med intestinal type tumor hadde størst effekt av FLOT sammenlignet med ECF/ECX (23 % patologisk komplett respons vs. 10%). For pasienter med diffus type histologi var det kun 3 % (én pasient) med patologisk komplett respons i begge gruppene (Al-Batran et al., 2016). Fase III data i studien viste at effekten av FLOT på overlevelse var størst for ikke-diffus type (HR 0,75), men overlevelsen var også bedret ved diffus type (HR 0,85). For gruppen med signetringceller ble det også vist bedret overlevelse med FLOT (HR 0,74) (Al-Batran et al., 2019). Det anbefales at også diffuse adenokarsinom og signetringcelletumores vurderes for perioperativ eller adjuvant onkologisk behandling.
Anbefaling
- For pasienter med resektabel kreft i magesekken med cT2 svulst eller høyere og/eller N+ som er i god allmenntilstand, WHO/ECOG 0-1, anbefales neoadjuvant og adjuvant cytostatikabehandling med FLOT regimet (evidensgrad A).
- Alternativ: FOLFOX
- Adjuvant kjemoterapi (fluoropyrimidin alene eller i kombinasjon med oxaliplatin), kan vurderes til pasienter som er operert uten neoadjuvant behandling ved patologisk stadium Ib – III
- Postoperativ kombinert radio-kjemoterapi kan vurderes ved ufrie reseksjonsrender.
- Pasienter som har mottatt neoadjuvant radiokjemoterapi uten komplett patologisk respons, kan vurderes for adjuvant nivolumab i ett år.
Oppfølging og kontroll etter avsluttet kurativ behandling
Sist faglig oppdatert: 28.09.2021
Endoskopisk behandling
Etter endoskopisk reseksjon for tidligcancer bør første gastroskopikontroll være etter 3 måneder, så ved 6 og 12 måneder. Deretter årlig i totalt 5 år. Reseksjon med piecemeal teknikk innebærer større risiko for lokalt residiv (J. Y. Lee et al., 2012), Ved usikker histologisk reseksjonsrand gjøres endoskopikontroll allerede etter 6 uker.
Operativ behandling
Det er ikke vist at et strukturert kontrollopplegg etter kurativ operativ behandling for kreft i magesekken bedrer overlevelse (Allum et al., 2011; D'Ugo, Biondi, Tufo, & Persiani, 2013; Smyth et al., 2016).
Ernæringsproblemer med vekttap, anemi og mangel på vitaminer og mineraler er ikke uvanlig etter ventrikkelreseksjon eller gastrektomi selv om et fåtall utvikler alvorlige symptomer (96). En randomisert studie med 371 pasienter fra Kina (Meng et al., 2021), har vist en viss effekt av supplement med en daglig næringsdrikk i tre måneder etter total og subtotal gastrektomi. Pasientene som fikk ernæringsoppfølging og næringsdrikk versus ernæringsoppfølging alene, hadde signifikant mindre vekttap, mindre tap av skjelettmuskulatur og mindre kjemoterapitoksisitet. Effektene var ganske beskjedne og bør bekreftes i flere studier, men studien gir en viss støtte for forskrivning av næringsdrikk til pasienter etter ventrikkelkirurgi for kreft.
Vurdering og oppfølging ved ernæringsfysiolog bør inngå som del av postoperativ rutine, og pasientene skal tilbys kontroll med henblikk på disse forhold hos kirurg 6-8 uker postoperativt. Videre oppfølging kan skje i samarbeide med fastlege. Vit-B12 gis 3-4 ganger i året. Ved langvarig ernæringsproblem eller plager som kan gi mistanke om residiv eller metastaser som kan være gjenstand for palliativ behandling bør terskelen være lav for å tilby pasientene en vurdering ved kirurgisk poliklinikk.
Anbefaling
- Etter endoskopisk reseksjon anbefales at pasientene følges med regelmessige endoskopiske kontroller i fem år (evidensgrad D).
- Postoperativ kontroll er ikke indisert med tanke på å oppdage tilbakefall av sykdommen (evidensgrad D).
- Postoperativ kontroll er viktig for å sikre adekvat ernæringstilstand (evidensgrad D).
Seneffekter
Sist faglig oppdatert: 28.09.2021
Rapport om seneffekter etter kreftbehandling
Helsedirektoratet ga ut rapporten Seneffekter etter kreftbehandling i 2020 (Helsedirektoratet, 2020). Målgruppen for rapporten er klinikere og annet helsepersonell som er i kontakt med kreftpasienter og pasienter som har vært gjennom kreftbehandling. Rapporten har kapitler om sekundær kreft, kardiovaskulære seneffekter, pulmonale seneffekter, hormonforstyrrelser etter kreftbehandling, seksualitet etter kreftbehandling, kognitive problemer, fatigue, langtidsbivirkninger etter behandling av hjernesvulst, psykososiale forhold, tann- og munnhuleproblemer, og spesielle forhold etter kreftbehandling hos barn.
Det er særlig viktig at fastleger, som skal følge opp pasienter som er skrevet ut fra spesialisthelsetjenesten, er oppmerksomme på risikoen for seneffekter etter kreftbehandling, og at disse kan debutere lenge etter avsluttet behandling.
Behandling av metastaserende sykdom og Livsforlengende og palliativ behandling
Kirurgi som livsforlengende behandling ved langt fremskredet sykdom
Sist faglig oppdatert: 28.09.2021
Et svensk registerstudium av 7,559 svenske pasienter med kreft i magesekken viste at 50 % hadde spredning. 1,945 (26%) hadde spredning til et organsystem mens 980 (13%) hadde spredning til multiple sites. Vanligst metastatisk focus var lever (48%), peritoneum (32%), lunger (15%) og beinvev (12%). 857 pasienter (11%) hadde had spredning til lymfeknuter eller uspeisifiserte områder (Riihimäki, Hemminki, Sundquist, Sundquist, & Hemminki, 2016).
Signet ring adenocarcinomas var forbundet med metastaser til peritoneum, beinvev og ovarier. Mindre hyppig til lunge og lever. Cardia cancer var assosiert med spredning til lunge, nervesystemet og beinvev, mens peritoneale metastaser var hyppigere ved non-cardiale metastaser.
Metastaser i lever og peritoneum var oftere singel site metastaser og lungemetastaser var oftere assosiert med levermetastaser.
Non-cardiac og signet-celle cancere metastaserte oftere til peritoneum og sistnevnte også til beinvev og ovarier. 56 % av kvinner med ovarial metastaser hadde også peritoneale metastaser.
Metastatisk sykdom innebærer svært kort forventet levetid og effektiv behandling kan innebære betydelig levetidsgevinst.
Kirurgi som del av livsforlengende behandling ved langt fremskredet ventrikkelcancer er omstridt (Waddell et al., 2014). «Evidence level» er lavt og utgjøres i hovedsak av pasientserier og komparative studier. En fase III multisenter studie av pasienter med spredning (til lever (H1) / peritoneum / para-aortale lymfeknuter) ble stoppet etter interim analyse. Overlevelse blant pasienter behandlet med kirurgi etterfulgt av kjemoterapi var ikke forskjellig fra kjemoterapi alene (Fujitani et al., 2016). Men studien inkluderte flere typer spredning og den kirurgiske prosedyre omfattet ikke reseksjon av metastaser utover lymfeknutemetastaser nær ventrikkelen (D1).
Langt fremskrevet (T4b)
Voluminøs tumor eller cancer i ventrikkel som er fast mot nabostrukturer kan lett bli definert som ikke resektabel. Flere studier har vist at multivisceral reseksjon kan bidra til lokal kontroll og 5 års overlevelse på mellom 20 – 38 % i selekterte studier. Oppnådd R-stadium (R0) og utbredelse av lymfeknutemetastaser var signifikante prognostiske faktorer. Pasienter tilgjengelig for R0 stadium oppnådde 5 års overlevelse på 43.7 % mot 31.4 % ved R1 and 0 % ved R2 (p<0.001) (Pacelli et al., 2013). Selv palliative reseksjoner (R1/R2) kan gi overlevelse signifikant utover det som oppnås med «bypass» operasjoner og simpel eksplorasjon. Ingen pasienter med «bypass» operasjon eller eksplorative prosedyrerer kan forvente 5-års overlevelse (Lai et al., 2011).
Det er sammenheng mellom affisert naboorgan og langtidsoverlevelse. To og fem års overlevelse blant pasienter med invasjon av pankreas (42.6 og 23.3 %) er signifikant svakere enn to og fem års overlevelse blant pasienter i kombinert gruppe (mesocolon + lever) (57.5 and 42.1 %, p = 0.002). Invasjon i pancreas er nært knyttet til høyere lymfeknute stadium, flere andre organaffeksjoner og høyere postoperativ komplikasjonsfrekvens. Det var heller ingen langtids overlevere blant dem med pankreatoduodenektomi (Min et al., 2012).
Overlevelsesgevinst ved mulitvisceral reseksjon forekommer, men med økt risiko for komplikasjoner. Gastrektomi sammen med reseksjon av to eller flere organ øker komplikasjonsrisiko signifikant (RR 4.39, CI 1.36–16.1) (Martin, Jaques, Brennan, & Karpeh, 2002).
Levermetastaser
Grunnpilaren i behandling av levermetastaser fra ventrikkel cancer er palliativ kjemoterapi, og kun en liten andel av pasientene har blitt tilbudt leverkirurgi (0.47 – 2.35 %). Det er samtidig erfart at pasienter med solitære og metakrone levermetastaser synes å ha god nytte av leverkirurgi (Okano et al., 2002; Waddell et al., 2014) og interessen for kirurgisk behandilng av metakrone levermetastaser har vært økende.
Det er også demonstrert at reseksjon av synkrone metastaser har betydning for langtidsoverlevelse og at reseksjon av levermetastaser med diameter mindre enn 5 cm gir lengst overlevelse (Komeda et al., 2014; Ueda et al., 2009).
I en metaanalyse av observasjonsstudier ble det funnet synkrone levermetastaser hos 6 % (95 % CI: 5–7) og samlet synkrone og metakrone hos 14 % (95 % CI: 7–27) av pasientene. Pasienter som gjennomgikk aggressiv lokal behandling av levermetastaser hadde signifikant bedret overlevelse enn dem som kun fikk palliasjon eller systemisk behandling (HR: 0.54, 95 % CI 0.46–0.95). Palliativ lokal behandling av levermetastaser innebar også bedret overlevelsesrate sammenlignet med kirurgi uten leverkirurgi eller systemisk palliasjon ((HR 0.50, 95 % CI 0.26– 0.96) (Ambiru et al., 2001; Martella et al., 2015).
En annen metaanalyse bekreftet at kirurgisk reseksjon av levermetastaser er assosiert med signifikant bedring av overlevelse (HR: 0.50; 95 % CI 0.41–0.61; P<0.001). Kirurgisk behandling av solitære levermetastaser var assosiert med signifikant bedre 5 års overlevelse enn reseksjon av multiple levermetastaser (OR: 0.31; 95 % CI 0.13–0.76; P= 0.011). Det var ikke mulig å påvise forskjell i 5 års overlevelse etter behandling av synkrone og metakrone lever metastaser (OR: 1.28; 95 % CI 0.46–3.57; P: 0.631) (Markar et al., 2016).
Reseksjon av solitær/få metastaser i lever kan vurderes i MDT.
Bimodal behandling av primært metastatisk ventrikkel kreft.
Perioperativ kjemoterapi med FLOT –regime til pasienter med begrenset metastatisk sykdom har vist å bidra til forlenget overlevelse AIO-FLOT3 (Al-Batran, Homann, et al., 2017).
I denne studien ble det utført reseksjon av metastaser sammen med radikal ventrikkel reseksjon (D2)
| Inkluder bar | Avgrenset til: |
| ||||
| ECOG 0 - 1 |
| |||||
| Operabel primærtumor |
| |||||
| Avgrenset metastatisk sykdom | Retroperitoneale lymfeknuter | Para-aortale | Inter aorto-cavale | Para-pancreatisk | Mesenteriale |
|
|
| Alene, og/ eller samtidig |
| ||||
|
|
Metastaser i ett av følgende organsystem | P1 carcinomatose | Lever < 5 metastaser | Lunge Unilateral | Krukenberg | Binyre Uni/bilaterale
|
| Ekstrabdominale lymfeknuter (supraclavikulære / cervicale | Beinmetastaser innen ett strålefelt | Andre avgrensede metastaser |
|
| ||
Det var relativt få pasienter i kirurgi gruppen (n = 60) og de fordelte seg med retroperitoneale lymfeknuter (n = 27 patients / 45%), lever metastaser (n=11, 18.3%), lungemetastaser (n=10, 16.7%), lokal peritoneale metastaser (n =4, 6.7%), og andre foci (n=8, 13.3%). 36 av pasientene (60%) ble operert og median overlevelse ble oppnådd ved 31.3 måneder (95% CI, 18.9- øvre nivå ikke oppnådd) i kirurgigruppen og 15.9 måneder (95% CI, 7.1-22.9) blant ikke opererte.
Oppsettet i FLOT 3 prøves ut i en fase III studie hvor kjemoterapi alene sammenlignes med 4 – 8 cykler perioperativ kjemoterapi hos pasienter med begrenset metastatisk sykdom. NCT02578368 / FLOT-5 (Al-Batran, Goetze, et al., 2017). Inklusjon av pasienter pågår etter skissen i tabell fra FLOT 3. Inklusjon pågår frem til desember 2021.
Kirurgi ved palliative inngrep
Sist faglig oppdatert: 28.09.2021
Ved obstruksjon må reseksjon, gastrojejunostomi eller stent vurderes (Jeurnink et al., 2007). Gastrektomi kan gi god palliasjon i utvalgte tilfeller hos unge personer med forventet levetid over fire - fem måneder (Haugstvedt, Viste, Eide, & Söreide, 1989) (evidensnivå3). Ved fjernmetastaser uten stenosesymptomer er reseksjon ikke indisert. Stent er særlig aktuelt ved stenose i distale del av ventrikkel og pylorus og kan føre til god symptomlindring (Larssen et al., 2011).
Strålebehandling
Sist faglig oppdatert: 28.09.2021
Ved lokale symptomer fra primærtumor, for eksempel smerter, blødning og obstruksjon, kan palliativ strålebehandling være aktuelt. Man kan gi ekstern strålebehandling, f.eks 3 Gy x 10-12 til totalt 30-36 Gy (M. M. Kim et al., 2008), alternativt 2.5 Gy x 14 til 35 Gy (Tey et al., 2007), (evidensnivå 3). Strålebehandling i denne regionen er imidlertid forbundet med til dels betydelige bivirkninger.
Medikamentell behandling ved avansert eller metastatisk sykdom
Sist faglig oppdatert: 28.09.2021
Førstelinjebehandling
Kreft i magesekken med fjernmetastaser og/eller ikke-resektabel svulst er vanligvis ikke mulig å helbrede, og intensjonen for all behandling blir derfor palliativ. Hensikten med systemisk behandling er å stabilisere sykdommen, lindre plager, forlenge symptomfattig periode og helst også øke levetiden. I en Cochrane-review er det vist at kombinasjonskjemoterapi bedrer både livskvalitet og overlevelse framfor monoterapi, men har økt toksisitet (Wagner et al., 2010). Det er rapportert responsrater på 17-51 % og median overlevelse på 6-11 måneder.
Det er vist overlevelsesgevinst ved cellegiftbehandling både i 1.- og 2.- og 3.-linjebehandling sammenlignet med ”best supportive care”. Flere ulike cytostatikagrupper er vist å ha effekt, og ved valg av terapi er det derfor viktig å se både på selve behandlingseffekten og på bivirkningsprofilen. Pasientens komorbiditet og funksjonsklasse må alltid tas med i vurderingen ved valg av behandling. Vanligvis vil palliativ kjemoterapi hos pasienter med dårlig almenntilstand (ECOG status >2) gi liten eller ingen nytte, men må vurderes individuelt.
Pasienter i god allmenntilstand og ECOG 0-2 bør vurderes for palliativ kjemoterapibehandling. For de fleste pasientene anbefales tostoffs-regimer med fluoropyrimidine (fluorouracil eller capecitabin) og platinum (oxaliplatin eller cisplatin). Oxaliplatin foretrekkes ofte fremfor cisplatin grunnet mindre toksisitet. Aktuelle regimer er CapOx, FOLFOX eller FLOX.
Trestoffs-regimer har en ikke ubetydelig toksisitet og bør reserveres til yngre pasienter i god allmenntilstand og ECOG 0-1 med hyppig vurdering av toksisitet.Trestoffs-kombinasjonen av cisplatin, 5FU og epirubicin (ECF) var tidligere ansett som standard førstelinjebehandling i Europa og Norge. REAL2 studien fra 2008 viste at oxaliplatin i stedet for cisplatin og capecitabine i stedet for iv 5FU (EOX) var like effektivt i kombinasjon med epirubicin. Totaloverlevelse(OS) i EOX-gruppen i denne studien var 11,2 måneder (Cunningham et al., 2008). I palliativ kjemoterapi har dublet og triplet regimer med platinum/5-FU +/- epirubicin vist responsrater mellom 35-45 % (Cunningham et al., 2008). Man har også sett lik effekt av oxaliplatin i kombinasjon med 5-FU sammenlignet med ECF (Enzinger et al., 2010). Disse resultatene har også gjort at nytten av epirubicin har blitt diskutert. En tidligere metaanalyse fra 2010 viste signifikant overlevelsesgevinst ved tillegg av antracycliner til cisplatin og 5-FU (Ter Veer et al., 2016; Wagner et al., 2010), men en nyere meta-analyse konkluderer med manglende tilleggsgevinst av antracyclin (Carmona-Bayonas et al., 2018; Ter Veer et al., 2016).
Docetaxel i kombinasjon med cisplatin og 5FU (DCF) har vist en noe bedre OS enn CF alene (OS 9,2 mnd vs 8,6 mnd), men høyere toksisitet i taxangruppen gjør denne kombinasjonen mindre aktuell (Van Cutsem et al., 2006). En senere studie med modifisert DCF har antydet at man kan oppnå minst like gode resultater ved å doseredusere, og samtidig oppnå langt lavere toksisitet (Shah et al., 2015).
Det mest lovende trestoffs-regimet var vurdert å være kombinasjon av fluoropyrimidin, oxaliplatin og et taxan (Ter Veer et al., 2016). FLOT-regimet (5-FU, oxaliplatin og docetaxel) har i to mindre studier vist lovende resultater med PFS på hhv 5,1 og 7,7, mnd., og OS på 11 og 14,6 mnd (Al-Batran et al., 2008; Van Cutsem et al., 2015). I tillegg viste den tyske randomisert fase 2/3 studie (FLOT4-AIO) som sammenliknet neoadjuvant kjemoterapi med FLOT regimet med ECF/ECX høyere histologisk komplett responsrate i resektatet ved FLOT regimet enn for ECF/ECX (16 % vs 6 %) (Al-Batran et al., 2019). FLOT vurderes nå å være det mest aktuelle trestoffs-regime
Pasienter som får et capecitabin-inneholdende regime, bør sannsynligvis ikke ta protonpumpehemmere samtidig. Det er reist bekymringer for at høyere pH-nivåer i magen kan hemme oppløsning og absorpsjon av capecitabin, noe som har negativ innvirkning på effekten (Chu et al., 2017; Sun et al., 2016).
Testing for eventuell DPD-mangel
Omtrent 3-8% av pasienter har partiell DPD-mangel (Dihydropyrimidin-dehydrogenase), og kan risikere alvorlig toksisitet av 5-FU og relaterte medikamenter. European Medicines Agency’s (EMA’s) anbefalte nylig at alle pasienter som skal motta 5-FU, kapecitabine eller tegafur bør testes for eventuell mangel på DPD før man starter slik behandling. DPYD testing etableres nå i Norge og pasienter bør da testes før oppstart av 5-FU basert behandling. Det anbefales test for enten DPYD genotype (gentest) eller indirekte funksjonstest av DPD aktivitet (måling av endogene uracil-metabolitter i plasma). Ved genotype som tyder på redusert DPD-aktivitet, bør en følge internasjonale retningslinjer med redusert startdose (Amstutz et al., 2018). Ved genotype som tyder på manglende DPD-aktivitet, bør behandling med 5-FU, kapecitabine og tegafur unngås.
Trastuzumab
ToGa-studien, en randomisert fase III-studie, undersøkte effekten av trastuzumab i kombinasjon med kjemoterapi i 1. linje ved HER2-positive svulster i gastroøsofageale overgang og magesekk (ikke resektabel eller metastatisk sykdom). Det ble påvist en signifikant bedret median OS i favør av trastuzumab-gruppen (13,8 vs 11.1 mnd) (Bang et al., 2010). Pasienter med høyest ekspresjon av HER2 (IHC 2+ og FISH+, eller IHC 3+) hadde størst gevinst av tilleggsbehandling med trastuzumab med bedring av median overlevelse fra 11.8 mnd. til 16 mnd. og ORR var 47 % vs 35 %. Slik tilleggsbehandling er derfor standardbehandling i avansert/metastatisk sykdom. Den optimale kjemoterapiregime for pasienter som får trastuzumab er ikke etablert. Vi anbefaler cisplatin/fluoropyrimidin eller oksaliplatin/fluoropyrimidin regimer (Ter Veer, Creemers, de Waal, van Oijen, & van Laarhoven, 2018).
Immunterapi i førstelinje
I KEYNOTE-062, en fase III-studie, var pembrolizumab monoterapi i første linje non-inferior og med signifikant mindre toksisitet enn kjemoterapi hos pasienter med CPS ≥1 (10,6 mot 11,1 måneder, HR 0,91). I subgruppeanalyse forbedret pembrolizumab total overlevelse (OS) sammenlignet med standard kjemoterapi hos pasienter med PD-L1 CPS≥10 (17,4 mot 10,8 måneder, HR, 0,69) (Shitara et al., 2020).
Nylig ble CheckMate-649 studien publisert, en fase III-studie som undersøkte kombinasjonsbehandling med nivolumab og cellegift (FOLFOX eller XELOX) versus kjemoterapi alene i 1. linje hos pasienter med avansert gastro-øsofagal adenokarsinom (Janjigian et al., 2021). Pasientene ble registrert i studien uavhengig av PD-L1 status. Primære endepunkter var total overlevelse og progresjonsfri overlevelse hos pasienter med PD-L1 CPS ≥ 5. Nivolumab med kjemoterapi viste en median OS på 14,4 måneder versus 11,1 måneder i kontrollgruppen for subpopulasjonen PD-L1 CPS ≥ 5 (HR 0,71 [0,59-0,86], p <0,0001). Gevinst med nivolumab ble også funnet ved PD-L1 CPS ≥ 1 og i hele kohorten, men den var noe mindre uttalt (hhv. 14 vs. 11,3 måneder og 13,8 mot 11,6 måneder). Alle forhåndsdefinerte undergrupper hadde fordel av nivolumab-behandling. Det synes å være spesielt god effekt av kombinasjonen Nivolumab og kjemoterapi ved MSI svulster. I denne studien var median overlevelse for denne pasientgruppen 8,8 mndr. ved kjemoterapi alene, mens median overlevelse ikke er nådd ved kombinasjonen. Det var ingen ekstra sikkerhetssignaler fra studien, og kombinasjonsterapi ble godt tolerert.
Resultatene tyder på at nivolumab kombinert med kjemoterapi kan bli standard førstelinje palliativ behandling for avanserte gastro-øsofagale adenokarsinom, endelig publikasjon avventes.
Lokalavansert ikke-resektabel kreft i magesekken uten metastaser
Optimal behandling av lokalavansert ikke-resektabel kreft i magesekken uten fjernmetastaser er ikke tilstrekkelig kartlagt. De fleste vil i slike tilfeller anbefale snarest mulig oppstart av kjemoterapi tilsvarende førstelinje palliativ kjemoterapi som oftest vil inkludere platinum, fluoropyrimidin +/- taxan. For primært ikke-resektable svulster, kan et regime med høyere respons-rate være ønskelig, og kombinasjonsregimer som har vist høyere responsrater i kliniske studier, bør kunne vurderes ut fra antatt toleranse hos pasienten. For pasienter med primært inoperabel HER-2 positiv sykdom, vil man kunne vurdere å legge trastuzumab til dublet kjemoterapi (platinum og 5-FU) siden dette har vist en høyere responsrate ved avansert / metastatisk sykdom. FLOT regimet vil være et egnet regime i neoadjuvant behandling av potensielt resektabel HER-2 negativ svulst. Ved respons på behandling, bør pasienter med primært lokalavanserte ikke-resektable tumores vurderes på ny om kirurgi anbefales eller ikke.
Andrelinje eller senere behandling
Irinotecan har vist øket overlevelse ved 2.-linjebehandling sammenlignet med best supportive care. Docetaxel viste i COUGAR-02-studien en OS på 5,2 mnd mot 3,6 mnd med best supportive care. En fase III-studie viste ingen signifikant forskjell for OS mellom paclitaxel og irinotecan for pasienter tidligere behandlet med 5-FU og platinum-basert cellegift (Ford et al., 2014; Fuchs et al., 2014; Hironaka et al., 2013; Wilke et al., 2014).
Flere enkeltstoffer og kombinasjoner har vist effekt på OS i 2.-linjebehandling. Ramucirumab er et monoklonalt antistoff som hemmer angiogenese via VGFR-2. Det foreligger to randomiserte studier hvor ramucirumab viser effekt på OS i 2.-linjebehandling. I REGARD-studien viste ramucirumab i monoterapi en OS på 5,2 mnd mot 3,8 mnd for placebogruppen. RAINBOW-studien sammenlignet paclitaxel alene med paclitaxel og ramucirumab i 2.-linje. Her viste kombinasjonsarmen en OS på 9,6 mnd mot 7,4 mnd for paclitaxel alene.
I flere studier blir det rapportert at noen av pasientene får tredjelinjebehandling. I en randomisert fase III-studie av pasienter med kjemo-refraktær kreft i magesekken (pasient behandlet med minst to tidligerelinjer kjemoterapi), forbedret trifluridin / tipiracil total overlevelse (OS) sammenlignet med placebo (OS 5.7 mot 3,6 måneders, hazard ratio (HR) 0,69, P = 0,00058) (Shitara, Doi, et al., 2018). Trifluridin/tipiracil kan vurderes i tredjelinje for en selektert pasientgruppe i god funksjonsklasse (ECOG 0-I), men den har foreløpig ikke vært oppe til behandling i Beslutningsforum. Det er for øvrige cellegifter ikke dokumentert at tredjelinjebehandling har bedre effekt enn beste lindrende behandling i en vestlig populasjon.
Immunterapi ved 2. og senere linje
Det er dokumentert gode resultater med immunterapi ved bruk av antistoffene pembrolizumab eller nivolumab rettet mot PD-1 ved metastaserende gastro-øsofagal adenokarsinom (mGØA).
I KEYNOTE-061 fase III-studien (n = 592), ble pembrolizumab sammenlignet med paclitaxel i andrelinje ved mGØA. Pembrolizumab ga ikke signifikant bedret overlevelse sammenlignet med paclitaxel med PD-L1 CPS 1 eller høyere. I subgruppeanalyse var effekten av Pembrolizumab større for pasienter med PD-L1 CPS≥10 (HR 0,64; 95 % CI 0,41–1,02, median OS 10,4 måneder vs. 8 måneder) (Shitara, Özgüroğlu, et al., 2018).
I KEYNOTE-059 fase II-studien, kohort 1 undersøkte pembrolizumab monoterapi ved mGØA hos 259 pasienter i 3. eller senere behandlingslinje. Den objektive responsraten (ORR) og sykdomskontrollraten (DCR) var henholdsvis 12 % og 27 %, og median OS var 5,6 måneder. ORR hadde en tendens til å være høyere i PD-L1-positiv (Combined Positive Score (CPS) ≥ 1) og MSI-svulster (Shitara, Özgüroğlu, et al., 2018).
I en koreansk fase II-studie, ble 61 pasienter med mGØA behandlet med pembrolizumab i 2. eller 3. linje. Hos pasienter med MSI eller Epstein Barr virus (EBV) positive svulster, var det dramatisk respons (ORR var 85,7 % i MSI og 100 % i EBV positive). For 55 PD-L1 positive pasienter (CPS ≥ 1 %), var ORR signifikant høyere (50.0 % mot 0.0 % for PD-L1 negative), P <0,001) (S. T. Kim et al., 2018).
I ATTRACTION-2 fase III studie (Japan, Sør-Korea, Taiwan) ble 493 pasienter med metastatisk eller ikke resektabel kreft i magesekk eller gastroøsofageal overgang randomisert (2:1) til behandling med Nivolumab eller placebo i 3. eller senere behandlingslinje. OS var signifikant lenger i nivolumabgruppen med median overlevelse 5,26 mndr mot 4,14 mndr i placebogruppen (HR 0,63, 95 % CI 0,51-0,78, p <0,0001). 12 mndr overlevelse var 26,2 % ved nivolumab og 10,9 % ved placebo (Kang et al., 2017).
Anbefalinger
- Ved obstruksjon må reseksjon, gastrojejunostomi eller stent vurderes. Gastrektomi kan vurderes i utvalgte tilfeller (evidensgrad C). Ved fjernmetastaser og primærsvulst uten stenosesymptomer er reseksjon ikke indisert.
- Ved lokale symptomer fra primærtumor bør strålebehandling vurderes.
- Pasienter i god almenntilstand og WHO 0-2 bør vurderes for cytostatikabehandling (evidensgrad A).
- Pasienter med primært lokalavansert ikke-resektabel tumor som respondere på behandling vurderes på nytt hvorvidt kirurgi anbefales eller ikke.
- For pasienter under 75 år, i god almenntilstand og WHO 0-1 anbefales
Førstelinje:
- FOLFOX, CapOx , annet platinum/5-FU, alternativt FLOT regimet
- Ved HER-2 positiv sykdom (IHC 2+ og FISH+, eller IHC3+) anbefales trastuzumab i kombinasjon med platinum / fluoropyrimidin doblet kjemoterapi.
- Ved PD-L1 CPS≥5 kan kombinasjonen av kjemoterapi og PD-1 hemmer vurderes.
- For de med PD-L1 CPS≥5 og begrenset metastatisk sykdom kan også PD-1 hemmer i monoterapi vurderes.
Andre og ytterligere linjebehandling:
- Taksaner (paclitaxel eller docetaxel) hvis ikke brukt før eller irinotecan basert (FOLFIRI, FLIRI eller irinotekan monoterapi)
- Tredjelinje/ fjerdelinje kjemoterapi med trifluridin / tipiracil kan vurderes for pasienter med ECOG 0–1.
- Pasienter som er ECOG 0-1(2) og PD-L1 CPS ≥ 10, MSI eller EBV positive kan immunterapi med PD-1 hemmer vurderes, etter progresjon på kjemoterapi
- For pasienter over 75 år eller yngre pasienter med redusert almenntilstand anbefales FLV, capecitabin monoterapi, irinotecan monoterapi, taksaner, evt FLOX/FOLFOX i redusert dose-regimet (evidensgrad D).
Supplerende behandling
Sist faglig oppdatert: 28.09.2021
Supplerende behandling kan være andre palliative tiltak; ernæring, fysisk aktivitet, psykososiale tiltak. Det vises til Nasjonalt handlingsprogram for palliasjon (Helsedirektoratet, 2019)(http://www.helsedirektoratet.no/publikasjoner/nasjonalt-handlingsprogram-med-retningslinjer-for-palliasjon-i-kreftomsorgen-/Sider/default.aspx).
Prosess og metode for utarbeiding av retningslinjene
Hva er nasjonale retningslinjer
Sist faglig oppdatert: 28.09.2021
I henhold til Nasjonal helseplan (2007–2010) (Nasjonal helseplan (2007-2010), 2006) har Helsedirektoratet en normerende rolle for helsetjenesten på tvers av helseregioner og tjenestenivå. Helsedirektoratet har en koordinerende rolle for å utvikle overordnede referanserammer for kreftomsorgen, sammen med de regionale helseforetakene, kommunene og andre relevante myndighetsorganer og tjenester.
Lov om kommunale helse- og omsorgstjenester § 12‑5 (Nasjonale faglige retningslinjer, veiledere og kvalitetsindikatorer. § 12-5 I: Lov om kommunale helse- og omsorgstjenester m.m. (helse- og omsorgstjenesteloven)) fastslår at Helsedirektoratet er eneste aktør med mandat til å utvikle, formidle og vedlikeholde nasjonale faglige retningslinjer og veiledere som understøtter de mål som er satt for helse- og omsorgstjenesten.
De nasjonale faglige retningslinjene inneholder systematisk utviklede faglige anbefalinger som etablerer en nasjonal standard for utredning, behandling og oppfølging av pasientgrupper, diagnosegrupper og brukergrupper. Nasjonale faglige retningslinjer er et virkemiddel for å bidra til at helse- og omsorgstjenestene:
- Har god kvalitet
- Gjør riktige prioriteringer
- Ikke har uønsket variasjon i tjenestetilbudet
- Løser samhandlingsutfordringer
- Tilbyr helhetlige pasientforløp
Nasjonale faglige retningslinjer gir uttrykk for hva som anses som god praksis på utgivelsestidspunktet. Sentrale fagmiljøer og tjenestemottakere er aktivt involvert i utarbeidelsen.
Helsepersonell og alle deler av helse- og omsorgstjenesten er forpliktet til å yte forsvarlig helsehjelp. Retningslinjer, anerkjent fagkunnskap og allmenngyldige samfunnsetiske normer inngår som aksepterte grunnlag for vurdering av hva som er faglig forsvarlig. Retningslinjer er ment som et hjelpemiddel ved avveiningene tjenesteyterne må gjøre for å oppnå forsvarlig og god kvalitet i tjenesten. De er ikke rettslig bindende, men faglig normerende for valg man anser fremmer kvalitet, god praksis og likhet i tjenesten på utgivelsestidspunktet. Helsepersonell må likevel vise faglig skjønn i vurderingen av hver enkelt pasient for å ta hensyn til individuelle behov. Dersom helsepersonell eller institusjoner velger å fravike anbefalinger i en retningslinje, skal dette dokumenteres og begrunnes.
Helsetjenestens eiere og ledelse har ansvar for tilrettelegging av virksomheten slik at anbefalinger gitt i nasjonale faglige retningslinjer kan følges.
Kunnskapsbasert prosess
Sist faglig oppdatert: 28.09.2021
Helsedirektoratet legger til grunn at alle nasjonale retningslinjer skal være utarbeidet etter en metode med vekt på forskningsbasert kunnskap, tydelig og tilgjengelig dokumentasjon, brukermedvirkning, tverrfaglighet, fokus på praksis, implementering og oppdatering.
Det er en omfattende prosess å lage gode retningslinjer som tilfredsstiller krav til prosess, metode og transparens som er det nivå Helsedirektoratet og andre internasjonale organisasjoner har lagt til grunn for utforming av anbefalinger.
Helsedirektoratet ønsker i arbeidet med nasjonale retningslinjer for kreftbehandling å bygge på det arbeid faggruppene tilsluttet Onkologisk Forum i en årrekke har gjort med å lage behandlingsveiledere.
Gradering av kunnskapsgrunnlaget
Sist faglig oppdatert: 28.09.2021
| Studietype | Evidensnivå | Gradering av evidensnivå |
|---|---|---|
| Kunnskap som bygger på systematiske oversikter og meta-analyser av randomiserte kontrollerte studier. Kunnskap som bygger på minst én randomisert kontrollert studie | Nivå 1a
Nivå 1b | A |
| Kunnskap som bygger på minst én godt utformet kontrollert studie uten randomisering Kunnskap som bygger på minst én annen godt utformet kvasi-eksperimentell studie uten randomisering | Nivå 2a Nivå 2b | B |
| Kunnskap som bygger på godt utformede ikke eksperimentelle beskrivende studier, som sammenlignende studier, korrelasjonsstudier og case studier | Nivå 3 | C |
| Kunnskap som bygger på rapporter eller oppfatninger fra eksperter, komiteer og/eller klinisk ekspertise hos respekterte autoriteter | Nivå 4 | D |
Ved utarbeiding av nasjonale retningslinjer stilles det krav om at all relevant kunnskap på området hentes frem, beskrives og vurderes på en systematisk og åpen måte.
I denne retningslinjen har man benyttet følgende graderingsmodell for å vise hvilket vitenskapelig grunnlag kunnskapen er basert på.
I disse retningslinjene er kun kunnskapsgrunnlaget og ikke anbefalingene gradert. Hvis selve anbefalingene skal graderes må man, i tillegg til å ha vurdert kunnskapsgrunnlaget, også legge inn en vurdering av både kost-nytte og andre forhold (klinisk erfaring, skjønn, etikk, osv). Dette er ikke gjort eksplisitt i forbindelse med dette arbeidet, og anbefalingene er derfor ikke gradert.
Bakgrunn og arbeidsprosess
Sist faglig oppdatert: 28.09.2021
Det nasjonale handlingsprogrammet med retningslinjer for diagnostikk, behandling og oppfølging av pasienter med kreft i magesekken ble første gang publisert 20. desember 2007.
Forfattere av det første handlingsprogrammet var følgende fagpersoner:
- Overlege, dr.med. Gunilla Frykholm, Onkologisk avdeling, St. Olavs Hospital
- Overlege, prof. Asgaut Viste, Kirurgisk avdeling, Haukeland Universitetssykehus, Helse-Bergen
- Overlege, prof. Egil Johnsson, Gastrokir.avd., Ullevål universitetssykehus
- Seksjonsleder Marianne Gjertsen, Nasjonalt kunnskapssenter for helsetjenesten, har bistått fagpersonene med gradering av kunnskapsgrunnlaget.
Arbeidet med første revisjon av Nasjonalt handlingsprogram for kreft i magesekken ble utført av Norsk Gastrointestinal Cancer Gruppe (NGICG) sitt fagråd for øsofagus- og ventrikkelcancer (NGICG-ØV) på oppdrag fra Helsedirektoratet.
Arbeidsgruppe ved femte utgave av handlingsprogrammet (utgitt 15.juni 2018)
- Overlege, professor Egil Johnson, Avdeling for gastroenterologisk kirurgi, Oslo universitetssykehus
- Overlege Ghazwan Al-Haidari, Avdeling for kreftbehandling, Oslo universitetssykehus
- Overlege dr. med Truls Hauge, Gastromedisinsk avdeling, Oslo universitetssykehus
- Overlege dr.med. Geir Olav Hjortland, Avdeling for kreftbehandling, Oslo universitetssykehus.
- Overlege Else Marit Løberg, Avdeling for patologi, Oslo universitetssykehus, Ullevål
- Overlege Gjermund Johnsen, Gastrokirurgisk avdeling, St. Olavs Hospital
- Overlege Ingunn Hatlevoll, Kreftklinikken, St. Olavs Hospital
- Overlege Henning Hellan, Klinikk for bildediagnostikk, St. Olavs Hospital
- Overlege Eirik Kjus Aahlin, Gastrokirurgisk avdeling, Universitetssykehuset i Nord-Norge
- Overlege Helge Stenvold, Kreftavdelingen, Universitetssykehuset i Nord-Norge
- Overlege, professor Kjell Kåre Øvrebø, Avdeling for gastro- og akuttkirurgi, Haukeland universitetssykehus
- Overlege Bente Kristin Abelseth, Kreftavdelingen, Haukeland universitessykehus
- Liv Marit Dørum, seksjonsnestleder, fagansvarlig for kvalitetsregistrene, Kreftregisteret
- Siri Larønningen, spesialrådgiver, Kreftregisteret
- Ingunn Aune, spesialkonsulent, Kreftregisteret
Arbeidsgruppe ved sjette utgave av handlingsprogrammet (utgitt ……)
Helse Nord
- Eirik Kjus Aahlin, kirurg, Universitetssykehuset Nord-Norge Tromsø
- Helge Stenvold, onkolog, Universitetssykehuset Nord-Norge Tromsø
Helse Midt-Norge
- Gjermund Johnsen, kirurg, St. Olavs hospital
- Henning Hellan, radiolog, St. Olavs hospital
- Ingunn Hatlevoll, onkolog, St. Olavs hospital
Helse Vest
- Kjell Kåre Øvrebø, kirurg, Haukeland universitetssjukehus
- Bente Kristin Abelseth, onkolog, Haukeland universitetssjukehus
Helse Sør-Øst
- Egil Johnson, leder av fagrådet, kirurg, Oslo universitetssykehus Ullevål
- Geir Olav Hjortland, onkolog, Oslo universitetssykehus Ullevål
- Truls Hauge, gastromedisiner, Oslo universitetssykehus Ullevål
- Ghazwan Al-Haidari, onkolog, Oslo universitetssykehus Ullevål
- Else Marit Løberg, patolog, Oslo universitetssykehus Ullevål
For Kreftregisteret
- Inger Kristin Larsen, ledelsesrepresentant, seksjonsnestleder, forsker, Registeravdelingen
- Liv Marit Rønning Dørum, fagansvarlig, seksjonsnestleder, spesialrådgiver, Registerseksjonen
- Ingunn Aune, kvalitetsregisteransvarlig
Habilitet
Sist faglig oppdatert: 28.09.2021
Alle gruppens medlemmer ble i forbindelse med arbeidet bedt om å oppgi potensielle interessekonflikter. Ingen interessekonflikter ble oppgitt.
Helsedirektoratet har vurdert arbeidsgruppens medlemmer som habile i forhold til utarbeiding av utkast til nasjonale retningslinjer for diagnostikk, behandling, og oppfølging av kreft i magesekken.
Ressursmessige konsekvenser
Sist faglig oppdatert: 28.09.2021
De forslag som fremlegges ved disse retningslinjene vil ikke føre til økte kostnader for spesialisthelsetjenesten.
Oppdatering av retningslinjene
Sist faglig oppdatert: 28.09.2021
Utviklingen av ny behandling på kreftområdet er rask. Det kan være behov for å endre retningslinjene fordi det er behandling som er ”utdatert” eller at det er behov å starte en prosess med vurdering av aktuell ny og kostbar kreftbehandling.
Innholdet i Nasjonale retningslinjer for kreft i magesekken vil vurderes årlig og om nødvendig oppdateres.
Reviderte utgaver av handlingsprogrammet:
| Første utgave: | IS-1527 | Gjeldende i perioden 20.12.07 – 18.08.2013 |
| Andre utgave: | IS-2085 | Gjeldende i perioden 19.08.13 – 02.10.2014 |
| Tredje utgave: | IS-2224 | Gjeldende i perioden 03.10.14 – 16.08.2015 |
| Fjerde utgave: | IS-2361 | Gjeldende i perioden 17.08.15 – 14.06.2018 |
| Femte utgave: | IS-2643 | Gjeldende i perioden 15.06.18 – 27.09.2021 |
Referanser
Sist faglig oppdatert: 28.09.2021
Al-Batran, S. E., Goetze, T. O., Mueller, D. W., Vogel, A., Winkler, M., Lorenzen, S., . . . Moenig, S. P. (2017). The RENAISSANCE (AIO-FLOT5) trial: effect of chemotherapy alone vs. chemotherapy followed by surgical resection on survival and quality of life in patients with limited-metastatic adenocarcinoma of the stomach or esophagogastric junction - a phase III trial of the German AIO/CAO-V/CAOGI. BMC Cancer, 17(1), 893. https://doi.org/10.1186/s12885-017-3918-9
Al-Batran, S. E., Hartmann, J. T., Hofheinz, R., Homann, N., Rethwisch, V., Probst, S., . . . Jäger, E. (2008). Biweekly fluorouracil, leucovorin, oxaliplatin, and docetaxel (FLOT) for patients with metastatic adenocarcinoma of the stomach or esophagogastric junction: a phase II trial of the Arbeitsgemeinschaft Internistische Onkologie. Ann Oncol, 19(11), 1882-1887. https://doi.org/10.1093/annonc/mdn403
Al-Batran, S. E., Hofheinz, R. D., Pauligk, C., Kopp, H. G., Haag, G. M., Luley, K. B., . . . Tannapfel, A. (2016). Histopathological regression after neoadjuvant docetaxel, oxaliplatin, fluorouracil, and leucovorin versus epirubicin, cisplatin, and fluorouracil or capecitabine in patients with resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4-AIO): results from the phase 2 part of a multicentre, open-label, randomised phase 2/3 trial. The Lancet. Oncology, 17(12), 1697-1708. https://doi.org/10.1016/s1470-2045(16)30531-9
Al-Batran, S. E., Homann, N., Pauligk, C., Goetze, T. O., Meiler, J., Kasper, S., . . . Hofheinz, R. D. (2019). Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): a randomised, phase 2/3 trial. Lancet (London, England), 393(10184), 1948-1957. https://doi.org/10.1016/s0140-6736(18)32557-1
Al-Batran, S. E., Homann, N., Pauligk, C., Illerhaus, G., Martens, U. M., Stoehlmacher, J., . . . Hofheinz, R. D. (2017). Effect of Neoadjuvant Chemotherapy Followed by Surgical Resection on Survival in Patients With Limited Metastatic Gastric or Gastroesophageal Junction Cancer: The AIO-FLOT3 Trial. JAMA Oncol, 3(9), 1237-1244. https://doi.org/10.1001/jamaoncol.2017.0515
Allum, W. H., Blazeby, J. M., Griffin, S. M., Cunningham, D., Jankowski, J. A., & Wong, R. (2011). Guidelines for the management of oesophageal and gastric cancer. Gut, 60(11), 1449-1472. https://doi.org/10.1136/gut.2010.228254
Ambiru, S., Miyazaki, M., Ito, H., Nakagawa, K., Shimizu, H., Yoshidome, H., . . . Nakajima, N. (2001). Benefits and limits of hepatic resection for gastric metastases. American journal of surgery, 181(3), 279-283.
Amstutz, U., Henricks, L. M., Offer, S. M., Barbarino, J., Schellens, J. H. M., Swen, J. J., . . . Schwab, M. (2018). Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin Pharmacol Ther, 103(2), 210-216. https://doi.org/10.1002/cpt.911
Ba-Ssalamah, A., Matzek, W., Baroud, S., Bastati, N., Zacherl, J., Schoppmann, S. F., . . . Gore, R. M. (2011). Accuracy of hydro-multidetector row CT in the local T staging of oesophageal cancer compared to postoperative histopathological results. European radiology, 21(11), 2326-2335. https://doi.org/10.1007/s00330-011-2187-2
Bamias, A., Karina, M., Papakostas, P., Kostopoulos, I., Bobos, M., Vourli, G., . . . Fountzilas, G. (2010). A randomized phase III study of adjuvant platinum/docetaxel chemotherapy with or without radiation therapy in patients with gastric cancer. Cancer chemotherapy and pharmacology, 65(6), 1009-1021. https://doi.org/10.1007/s00280-010-1256-6
Bang, Y. J., Kim, Y. W., Yang, H. K., Chung, H. C., Park, Y. K., Lee, K. H., . . . Noh, S. H. (2012). Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet (London, England), 379(9813), 315-321. https://doi.org/10.1016/s0140-6736(11)61873-4
Bang, Y. J., Van Cutsem, E., Feyereislova, A., Chung, H. C., Shen, L., Sawaki, A., . . . Kang, Y. K. (2010). Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet (London, England), 376(9742), 687-697. https://doi.org/10.1016/s0140-6736(10)61121-x
Barry, J. D., Edwards, P., Lewis, W. G., Dhariwal, D., & Thomas, G. V. (2002). Special interest radiology improves the perceived preoperative stage of gastric cancer. Clinical radiology, 57(11), 984-988.
Best, L. M., Mughal, M., & Gurusamy, K. S. (2016). Laparoscopic versus open gastrectomy for gastric cancer. The Cochrane database of systematic reviews, 3, CD011389. https://doi.org/10.1002/14651858.CD011389.pub2
Bozzetti, F., Bonfanti, G., Bufalino, R., Menotti, V., Persano, S., Andreola, S., . . . Gennari, L. (1982). Adequacy of margins of resection in gastrectomy for cancer. Annals of surgery, 196(6), 685-690.
Brierley, J. D., Gospodarowicz, M. K., & Wittekind, C. (Eds.). (2017). TNM classification of malignant tumours (8. ed.). Chichester, West Sussex, UK ; Hoboken, NJ: John Wiley & Sons.
Bringeland, E. A., Wasmuth, H. H., Fougner, R., Mjones, P., & Gronbech, J. E. (2014). Impact of perioperative chemotherapy on oncological outcomes after gastric cancer surgery. The British journal of surgery, 101(13), 1712-1720. https://doi.org/10.1002/bjs.9650
Bringeland, E. A., Wasmuth, H. H., Johnsen, G., Johnsen, T. B., Juel, I. S., Mjones, P., . . . Gronbech, J. E. (2013). Outcomes among patients treated for gastric adenocarcinoma during the last decade. Journal of surgical oncology, 107(7), 752-757. https://doi.org/10.1002/jso.23320
Buckland, G., Travier, N., Huerta, J. M., Bueno-de-Mesquita, H. B., Siersema, P. D., Skeie, G., . . . Gonzalez, C. A. (2015). Healthy lifestyle index and risk of gastric adenocarcinoma in the EPIC cohort study. International journal of cancer, 137(3), 598-606. https://doi.org/10.1002/ijc.29411
Cancer Genome Atlas Research Network. (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature, 513(7517), 202-209. https://doi.org/10.1038/nature13480
Cancer Genome Atlas Research Network, Analysis Working Group: Asan University, BC Cancer Agency, Brigham and Women’s Hospital, Broad Institute, Brown University, . . . KU Leuven. (2017). Integrated genomic characterization of oesophageal carcinoma. Nature, 541(7636), 169-175. https://doi.org/10.1038/nature20805
Carmona-Bayonas, A., Jiménez-Fonseca, P., Custodio, A., Sánchez Cánovas, M., Hernández, R., Pericay, C., . . . Gallego, J. (2018). Anthracycline-based triplets do not improve the efficacy of platinum-fluoropyrimidine doublets in first-line treatment of advanced gastric cancer: real-world data from the AGAMEMON National Cancer Registry. Gastric cancer : official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association, 21(1), 96-105. https://doi.org/10.1007/s10120-017-0718-5
Cats, A., Jansen, E. P. M., van Grieken, N. C. T., Sikorska, K., Lind, P., Nordsmark, M., . . . Verheij, M. (2018). Chemotherapy versus chemoradiotherapy after surgery and preoperative chemotherapy for resectable gastric cancer (CRITICS): an international, open-label, randomised phase 3 trial. The Lancet. Oncology, 19(5), 616-628. https://doi.org/10.1016/s1470-2045(18)30132-3
Choi, Y. Y., Kim, H., Shin, S. J., Kim, H. Y., Lee, J., Yang, H. K., . . . Cheong, J. H. (2019). Microsatellite Instability and Programmed Cell Death-Ligand 1 Expression in Stage II/III Gastric Cancer: Post Hoc Analysis of the CLASSIC Randomized Controlled study. Annals of surgery, 270(2), 309-316. https://doi.org/10.1097/sla.0000000000002803
Chu, M. P., Hecht, J. R., Slamon, D., Wainberg, Z. A., Bang, Y. J., Hoff, P. M., . . . Sawyer, M. B. (2017). Association of Proton Pump Inhibitors and Capecitabine Efficacy in Advanced Gastroesophageal Cancer: Secondary Analysis of the TRIO-013/LOGiC Randomized Clinical Trial. JAMA Oncol, 3(6), 767-773. https://doi.org/10.1001/jamaoncol.2016.3358
Cui, M., Li, Z., Xing, J., Yao, Z., Liu, M., Chen, L., . . . Su, X. (2015). A prospective randomized clinical trial comparing D2 dissection in laparoscopic and open gastrectomy for gastric cancer. Medical oncology (Northwood, London, England), 32(10), 241. https://doi.org/10.1007/s12032-015-0680-1
Cunningham, D., Allum, W. H., Stenning, S. P., Thompson, J. N., Van de Velde, C. J., Nicolson, M., . . . Participants, M. T. (2006). Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. The New England journal of medicine, 355(1), 11-20. https://doi.org/10.1056/NEJMoa055531
Cunningham, D., Starling, N., Rao, S., Iveson, T., Nicolson, M., Coxon, F., . . . Norman, A. R. (2008). Capecitabine and oxaliplatin for advanced esophagogastric cancer. The New England journal of medicine, 358(1), 36-46. https://doi.org/10.1056/NEJMoa073149
D'Ugo, D., Biondi, A., Tufo, A., & Persiani, R. (2013). Follow-up: the evidence. Digestive surgery, 30(2), 159-168. https://doi.org/10.1159/000350878
de Boer, W. B., Ee, H., & Kumarasinghe, M. P. (2018). Neoplastic Lesions of Gastric Adenocarcinoma and Proximal Polyposis Syndrome (GAPPS) Are Gastric Phenotype. Am J Surg Pathol, 42(1), 1-8. https://doi.org/10.1097/pas.0000000000000924
den Hoed, C. M., & Kuipers, E. J. (2016). Gastric Cancer: How Can We Reduce the Incidence of this Disease? Current gastroenterology reports, 18(7), 34. https://doi.org/10.1007/s11894-016-0506-0
Diaz-Nieto, R., Orti-Rodriguez, R., & Winslet, M. (2013). Post-surgical chemotherapy versus surgery alone for resectable gastric cancer. The Cochrane database of systematic reviews, (9), CD008415. https://doi.org/10.1002/14651858.CD008415.pub2
Dikken, J. L., Jansen, E. P., Cats, A., Bakker, B., Hartgrink, H. H., Kranenbarg, E. M., . . . Verheij, M. (2010). Impact of the extent of surgery and postoperative chemoradiotherapy on recurrence patterns in gastric cancer. J Clin Oncol, 28(14), 2430-2436. https://doi.org/10.1200/jco.2009.26.9654
Dikken, J. L., van Sandick, J. W., Maurits Swellengrebel, H. A., Lind, P. A., Putter, H., Jansen, E. P., . . . Cats, A. (2011). Neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy for patients with resectable gastric cancer (CRITICS). BMC Cancer, 11, 329. https://doi.org/10.1186/1471-2407-11-329
Earle, C. C., & Maroun, J. A. (1998). Adjuvant chemotherapy after curative resection for gastric cancer: revisiting a meta-analysis of randomised trials. Proc Am Soc Clin Oncol 1998;17:263a. Proc Am Soc Clin Oncol, 17, 263a.
Enzinger, P. C., Burtness, B., Hollis, D., Niedzwiecki, D., Ilson, D., Benson, A. B., . . . Goldberg, R. M. (2010). CALGB 80403/ECOG 1206: A randomized phase II study of three standard chemotherapy regimens (ECF, IC, FOLFOX) plus cetuximab in metastatic esophageal and GE junction cancer. Journal of Clinical Oncology, 28(15 suppl), 4006. https://doi.org/10.1200/jco.2010.28.15_suppl.4006
Fitzgerald, R. C., Hardwick, R., Huntsman, D., Carneiro, F., Guilford, P., Blair, V., . . . Caldas, C. (2010). Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. Journal of medical genetics, 47(7), 436-444. https://doi.org/10.1136/jmg.2009.074237
Ford, H. E., Marshall, A., Bridgewater, J. A., Janowitz, T., Coxon, F. Y., Wadsley, J., . . . Dunn, J. A. (2014). Docetaxel versus active symptom control for refractory oesophagogastric adenocarcinoma (COUGAR-02): an open-label, phase 3 randomised controlled trial. The Lancet. Oncology, 15(1), 78-86. https://doi.org/10.1016/s1470-2045(13)70549-7
Forman, D., & Burley, V. J. (2006). Gastric cancer: global pattern of the disease and an overview of environmental risk factors. Best practice & research. Clinical gastroenterology, 20(4), 633-649. https://doi.org/10.1016/j.bpg.2006.04.008
Forskrift om prioritering av helsetjenester, rett til nødvendig helsehjelp fra spesialisthelsetjenesten, rett til behandling i utlandet og om klagenemnd (prioriteringsforskriften). FOR-2000-12-01-1208. Last update in FOR-2020-02-04-119 fra 01.03.2020. Retrieved from https://lovdata.no/dokument/SF/forskrift/2000-12-01-1208
Fuchs, C. S., Tomasek, J., Yong, C. J., Dumitru, F., Passalacqua, R., Goswami, C., . . . Tabernero, J. (2014). Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet (London, England), 383(9911), 31-39. https://doi.org/10.1016/s0140-6736(13)61719-5
Fujitani, K., Yang, H. K., Mizusawa, J., Kim, Y. W., Terashima, M., Han, S. U., . . . Tsujinaka, T. (2016). Gastrectomy plus chemotherapy versus chemotherapy alone for advanced gastric cancer with a single non-curable factor (REGATTA): a phase 3, randomised controlled trial. The Lancet. Oncology, 17(3), 309-318. https://doi.org/10.1016/s1470-2045(15)00553-7
Fukase, K., Kato, M., Kikuchi, S., Inoue, K., Uemura, N., Okamoto, S., . . . Asaka, M. (2008). Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: an open-label, randomised controlled trial. Lancet (London, England), 372(9636), 392-397. https://doi.org/10.1016/s0140-6736(08)61159-9
Gotoda, T., Yanagisawa, A., Sasako, M., Ono, H., Nakanishi, Y., Shimoda, T., & Kato, Y. (2000). Incidence of lymph node metastasis from early gastric cancer: estimation with a large number of cases at two large centers. Gastric cancer : official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association, 3(4), 219-225.
Hallinan, J. (2012). CT volumetry for gastric carcinoma staging. Poster presentert på European Congress of Radiology; Vienna, Austria 1-5 mars
Hallissey, M. T., Jewkes, A. J., Dunn, J. A., Ward, L., & Fielding, J. W. (1993). Resection-line involvement in gastric cancer: a continuing problem. The British journal of surgery, 80(11), 1418-1420.
Halm, E. A., Lee, C., & Chassin, M. R. (2002). Is volume related to outcome in health care? A systematic review and methodologic critique of the literature. Annals of internal medicine, 137(6), 511-520.
Haugstvedt, T., Viste, A., Eide, G. E., & Söreide, O. (1989). The survival benefit of resection in patients with advanced stomach cancer: the Norwegian multicenter experience. Norwegian Stomach Cancer Trial. World journal of surgery, 13(5), 617-621; discussion 621-612.
Helsedirektoratet. (2019). Nasjonalt handlingsprogram for palliasjon i kreftomsorgen (IS-2800). Oslo: Helsedirektoratet. Retrieved from https://www.helsedirektoratet.no/retningslinjer/palliasjon-i-kreftomsorgen-handlingsprogram
Helsedirektoratet. (2020). Seneffekter etter kreftbehandling (rev. ed.). Oslo: Helsedirektoratet. Retrieved from https://www.helsedirektoratet.no/rapporter/seneffekter-etter-kreftbehandling/Seneffekter%20etter%20kreftbehandling.pdf/_/attachment/inline/3d984c2a-7926-4d1a-a5f0-06d48fe7c95f:f3e498d059734ff34b013c1c206877e488e95600/Seneffekter%20etter%20kreftbehandling.pdf
Hironaka, S., Ueda, S., Yasui, H., Nishina, T., Tsuda, M., Tsumura, T., . . . Hyodo, I. (2013). Randomized, open-label, phase III study comparing irinotecan with paclitaxel in patients with advanced gastric cancer without severe peritoneal metastasis after failure of prior combination chemotherapy using fluoropyrimidine plus platinum: WJOG 4007 trial. J Clin Oncol, 31(35), 4438-4444. https://doi.org/10.1200/jco.2012.48.5805
Honório, M. X., Suzuki, C., Espin-Garcia, O., Allen, M. J., Ma, L., Almugbel, F. A., . . . Elimova, E. (2020). Asian-ethnicity related differences among patients with metastatic gastroesophageal junction (GEJ) and gastric adenocarcinoma. Annals of Oncology, 31(Suppl 4), S841-S873. http://dx.doi.org/10.1016/j.annonc.2020.08.1970
Hu, Y., Huang, C., Sun, Y., Su, X., Cao, H., Hu, J., . . . Li, G. (2016). Morbidity and Mortality of Laparoscopic Versus Open D2 Distal Gastrectomy for Advanced Gastric Cancer: A Randomized Controlled Trial. J Clin Oncol, 34(12), 1350-1357. https://doi.org/10.1200/jco.2015.63.7215
Ichikura, T., Ogawa, T., Chochi, K., Kawabata, T., Sugasawa, H., & Mochizuki, H. (2003). Minimum number of lymph nodes that should be examined for the International Union Against Cancer/American Joint Committee on Cancer TNM classification of gastric carcinoma. World journal of surgery, 27(3), 330-333. https://doi.org/10.1007/s00268-002-6730-9
Irino, T., Sano, T., Hiki, N., Ohashi, M., Nunobe, S., Kumagai, K., . . . Yamaguchi, T. (2018). Diagnostic staging laparoscopy in gastric cancer: a prospective cohort at a cancer institute in Japan. Surgical endoscopy, 32(1), 268-275. https://doi.org/10.1007/s00464-017-5673-z
Janjigian, Y. Y., Shitara, K., Moehler, M., Garrido, M., Salman, P., Shen, L., . . . Ajani, J. A. (2021). First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet (London, England), 398(10294), 27-40. https://doi.org/10.1016/s0140-6736(21)00797-2
Janssen, C. W., Jr., Lie, R. T., Maartmann-Moe, H., & Matre, R. (1991). The influence of age on the growth and spread of gastric carcinoma. British journal of cancer, 63(4), 623-625.
Japanese Gastric Cancer Association. (2017). Japanese gastric cancer treatment guidelines 2014 (ver. 4). Gastric cancer : official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association, 20(1), 1-19. https://doi.org/10.1007/s10120-016-0622-4
Jeurnink, S. M., Steyerberg, E. W., Hof, G., van Eijck, C. H., Kuipers, E. J., & Siersema, P. D. (2007). Gastrojejunostomy versus stent placement in patients with malignant gastric outlet obstruction: a comparison in 95 patients. Journal of surgical oncology, 96(5), 389-396. https://doi.org/10.1002/jso.20828
Kakeji, Y., Morita, M., & Maehara, Y. (2010). Strategies for treating liver metastasis from gastric cancer. Surgery today, 40(4), 287-294. https://doi.org/10.1007/s00595-009-4152-0
Kang, Y. K., Boku, N., Satoh, T., Ryu, M. H., Chao, Y., Kato, K., . . . Chen, L. T. (2017). Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (London, England), 390(10111), 2461-2471. https://doi.org/10.1016/s0140-6736(17)31827-5
Kawanaka, Y., Kitajima, K., Fukushima, K., Mouri, M., Doi, H., Oshima, T., . . . Hirota, S. (2016). Added value of pretreatment (18)F-FDG PET/CT for staging of advanced gastric cancer: Comparison with contrast-enhanced MDCT. Eur J Radiol, 85(5), 989-995. https://doi.org/10.1016/j.ejrad.2016.03.003
Kelly, R. J., Ajani, J. A., Kuzdzal, J., Zander, T., Van Cutsem, E., Piessen, G., . . . Moehler, M. (2021). Adjuvant Nivolumab in Resected Esophageal or Gastroesophageal Junction Cancer. The New England journal of medicine, 384(13), 1191-1203. https://doi.org/10.1056/NEJMoa2032125
Kelly, R. J., Ajani, J. A., Kuzdzal, J., Zander, T., Van Cutsem, E., Piessen, G., . . . Moehler, M. (2020). Adjuvant nivolumab in resected esophageal or gastroesophageal junction cancer (EC/GEJC) following neoadjuvant chemoradiation therapy (CRT): First results of the CheckMate 577 study. Annals of Oncology, 31(Suppl 4), S1142-S1215. /http://dx.doi.org/10.1016/j.annonc.2020.08.2299
Kelly, S., Harris, K. M., Berry, E., Hutton, J., Roderick, P., Cullingworth, J., . . . Smith, M. A. (2001). A systematic review of the staging performance of endoscopic ultrasound in gastro-oesophageal carcinoma. Gut, 49(4), 534-539.
Killeen, S. D., O'Sullivan, M. J., Coffey, J. C., Kirwan, W. O., & Redmond, H. P. (2005). Provider volume and outcomes for oncological procedures. The British journal of surgery, 92(4), 389-402. https://doi.org/10.1002/bjs.4954
Kim, D. J., Lee, J. H., & Kim, W. (2014). A comparison of total versus partial omentectomy for advanced gastric cancer in laparoscopic gastrectomy. World J Surg Oncol, 12, 64. https://doi.org/10.1186/1477-7819-12-64
Kim, J. J., Lee, J. H., Jung, H. Y., Lee, G. H., Cho, J. Y., Ryu, C. B., . . . Yoon, Y. B. (2007). EMR for early gastric cancer in Korea: a multicenter retrospective study. Gastrointestinal endoscopy, 66(4), 693-700. https://doi.org/10.1016/j.gie.2007.04.013
Kim, M. M., Rana, V., Janjan, N. A., Das, P., Phan, A. T., Delclos, M. E., . . . Krishnan, S. (2008). Clinical benefit of palliative radiation therapy in advanced gastric cancer. Acta oncologica (Stockholm, Sweden), 47(3), 421-427. https://doi.org/10.1080/02841860701621233
Kim, S. T., Cristescu, R., Bass, A. J., Kim, K. M., Odegaard, J. I., Kim, K., . . . Kang, W. K. (2018). Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med, 24(9), 1449-1458. https://doi.org/10.1038/s41591-018-0101-z
Kim, T. H., Park, S. R., Ryu, K. W., Kim, Y. W., Bae, J. M., Lee, J. H., . . . Kim, D. Y. (2012). Phase 3 trial of postoperative chemotherapy alone versus chemoradiation therapy in stage III-IV gastric cancer treated with R0 gastrectomy and D2 lymph node dissection. International journal of radiation oncology, biology, physics, 84(5), e585-592. https://doi.org/10.1016/j.ijrobp.2012.07.2378
Komeda, K., Hayashi, M., Kubo, S., Nagano, H., Nakai, T., Kaibori, M., . . . Uchiyama, K. (2014). High survival in patients operated for small isolated liver metastases from gastric cancer: a multi-institutional study. World journal of surgery, 38(10), 2692-2697. https://doi.org/10.1007/s00268-014-2608-x
Kondo, H., Gotoda, T., Ono, H., Oda, I., Yamaguchi, H., Saito, D., & Yoshida, S. (2001). Early gastric cancer: endoscopic mucosal resection. Annali italiani di chirurgia, 72(1), 27-31.
Kong, P., Cai, Q., Geng, Q., Wang, J., Lan, Y., Zhan, Y., & Xu, D. (2014). Vitamin intake reduce the risk of gastric cancer: meta-analysis and systematic review of randomized and observational studies. PLoS ONE [Electronic Resource], 9(12), e116060. https://doi.org/10.1371/journal.pone.0116060
Kurokawa, Y., Doki, Y., Mizusawa, J., Terashima, M., Katai, H., Yoshikawa, T., . . . Sasako, M. (2018). Bursectomy versus omentectomy alone for resectable gastric cancer (JCOG1001): a phase 3, open-label, randomised controlled trial. Lancet Gastroenterol Hepatol, 3(7), 460-468. https://doi.org/10.1016/s2468-1253(18)30090-6
Kwee, R. M., & Kwee, T. C. (2007). Imaging in local staging of gastric cancer: a systematic review. J Clin Oncol, 25(15), 2107-2116. https://doi.org/10.1200/jco.2006.09.5224
Kwon, H. C., Kim, M. C., Kim, K. H., Jang, J. S., Oh, S. Y., Kim, S. H., . . . Kim, H. J. (2010). Adjuvant chemoradiation versus chemotherapy in completely resected advanced gastric cancer with D2 nodal dissection. Asia-Pacific journal of clinical oncology, 6(4), 278-285. https://doi.org/10.1111/j.1743-7563.2010.01331.x
Lai, K. K., Fang, W. L., Wu, C. W., Huang, K. H., Chen, J. H., Lo, S. S., & Li, A. F. (2011). Surgical impact on gastric cancer with locoregional invasion. World journal of surgery, 35(11), 2479-2484. https://doi.org/10.1007/s00268-011-1246-9
Larsen, I. K. (Ed.). (2016). Cancer in Norway 2015: Cancer incidence, mortality, survival and prevalence in Norway. Oslo: Cancer Registry of Norway. Retrieved from https://www.kreftregisteret.no/globalassets/cancer-in-norway/2015/cin2015-special_issue-web.pdf
Larssen, L., Medhus, A. W., Hjermstad, M. J., Korner, H., Glomsaker, T., Søberg, T., . . . Hauge, T. (2011). Patient-reported outcomes in palliative gastrointestinal stenting: a Norwegian multicenter study. Surgical endoscopy, 25(10), 3162-3169. https://doi.org/10.1007/s00464-011-1680-7
Larsson, S. C., Bergkvist, L., & Wolk, A. (2006). Fruit and vegetable consumption and incidence of gastric cancer: a prospective study. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology, 15(10), 1998-2001. https://doi.org/10.1158/1055-9965.epi-06-0402
Lauren, P. (1965). The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta pathologica et microbiologica Scandinavica, 64, 31-49.
Lee, J. H., Lee, C. M., Son, S. Y., Ahn, S. H., Park, D. J., & Kim, H. H. (2014). Laparoscopic versus open gastrectomy for gastric cancer: long-term oncologic results. Surgery, 155(1), 154-164. https://doi.org/10.1016/j.surg.2013.06.015
Lee, J. Y., Choi, I. J., Cho, S. J., Kim, C. G., Kook, M. C., Lee, J. H., . . . Kim, Y. W. (2012). Routine follow-up biopsies after complete endoscopic resection for early gastric cancer may be unnecessary. Journal of gastric cancer, 12(2), 88-98. https://doi.org/10.5230/jgc.2012.12.2.88
Lee, S. M., Kim, S. H., Lee, J. M., Im, S. A., Bang, Y. J., Kim, W. H., . . . Choi, B. I. (2009). Usefulness of CT volumetry for primary gastric lesions in predicting pathologic response to neoadjuvant chemotherapy in advanced gastric cancer. Abdom Imaging, 34(4), 430-440. https://doi.org/10.1007/s00261-008-9420-8
Leong, T., Smithers, B. M., Michael, M., Gebski, V., Boussioutas, A., Miller, D., . . . Wong, R. (2015). TOPGEAR: a randomised phase III trial of perioperative ECF chemotherapy versus preoperative chemoradiation plus perioperative ECF chemotherapy for resectable gastric cancer (an international, intergroup trial of the AGITG/TROG/EORTC/NCIC CTG). BMC Cancer, 15, 532. https://doi.org/10.1186/s12885-015-1529-x
Liedman, B., Bosaeus, I., Hugosson, I., & Lundell, L. (1998). Long-term beneficial effects of a gastric reservoir on weight control after total gastrectomy: a study of potential mechanisms. The British journal of surgery, 85(4), 542-547. https://doi.org/10.1046/j.1365-2168.1998.00747.x
Lindahl, A. K., Harbitz, T. B., & Liavåg, I. (1988). The surgical treatment of gastric cancer: a retrospective study with special reference to total gastrectomy. European journal of surgical oncology : the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology, 14(1), 55-62.
Liu, T. S., Wang, Y., Chen, S. Y., & Sun, Y. H. (2008). An updated meta-analysis of adjuvant chemotherapy after curative resection for gastric cancer. European journal of surgical oncology : the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology, 34(11), 1208-1216. https://doi.org/10.1016/j.ejso.2008.02.002
Lov om pasient- og brukerrettigheter (pasient- og brukerrettighetsloven) § 2-2 Rett til vurdering. LOV-1999-07-02-63. Last update in LOV-2021-05-07-31 fra 01.07.2021. Retrieved from https://lovdata.no/lov/1999-07-02-63/§2-2
Macdonald, J. S., Smalley, S. R., Benedetti, J., Hundahl, S. A., Estes, N. C., Stemmermann, G. N., . . . Martenson, J. A. (2001). Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. The New England journal of medicine, 345(10), 725-730. https://doi.org/10.1056/NEJMoa010187
Mariette, C., Piessen, G., Briez, N., Gronnier, C., & Triboulet, J. P. (2011). Oesophagogastric junction adenocarcinoma: which therapeutic approach? The Lancet. Oncology, 12(3), 296-305. https://doi.org/10.1016/s1470-2045(10)70125-x
Markar, S. R., Mikhail, S., Malietzis, G., Athanasiou, T., Mariette, C., Sasako, M., & Hanna, G. B. (2016). Influence of Surgical Resection of Hepatic Metastases From Gastric Adenocarcinoma on Long-term Survival: Systematic Review and Pooled Analysis. Annals of surgery, 263(6), 1092-1101. https://doi.org/10.1097/sla.0000000000001542
Martella, L., Bertozzi, S., Londero, A. P., Steffan, A., De Paoli, P., & Bertola, G. (2015). Surgery for Liver Metastases From Gastric Cancer: A Meta-Analysis of Observational Studies. Medicine (Baltimore), 94(31), e1113. https://doi.org/10.1097/md.0000000000001113
Martin, R. C., 2nd, Jaques, D. P., Brennan, M. F., & Karpeh, M. (2002). Achieving RO resection for locally advanced gastric cancer: is it worth the risk of multiorgan resection? Journal of the American College of Surgeons, 194(5), 568-577.
Mayne, S. T., Risch, H. A., Dubrow, R., Chow, W. H., Gammon, M. D., Vaughan, T. L., . . . Fraumeni, J. F., Jr. (2001). Nutrient intake and risk of subtypes of esophageal and gastric cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology, 10(10), 1055-1062.
Meng, Q., Tan, S., Jiang, Y., Han, J., Xi, Q., Zhuang, Q., & Wu, G. (2021). Post-discharge oral nutritional supplements with dietary advice in patients at nutritional risk after surgery for gastric cancer: A randomized clinical trial. Clin Nutr, 40(1), 40-46. https://doi.org/10.1016/j.clnu.2020.04.043
Miki, Y., Tokunaga, M., Tanizawa, Y., Bando, E., Kawamura, T., & Terashima, M. (2015). Staging Laparoscopy for Patients with cM0, Type 4, and Large Type 3 Gastric Cancer. World journal of surgery, 39(11), 2742-2747. https://doi.org/10.1007/s00268-015-3144-z
Min, J. S., Jin, S. H., Park, S., Kim, S. B., Bang, H. Y., & Lee, J. I. (2012). Prognosis of curatively resected pT4b gastric cancer with respect to invaded organ type. Annals of surgical oncology, 19(2), 494-501. https://doi.org/10.1245/s10434-011-1987-6
Miyata, M., Yokoyama, Y., Okoyama, N., Joh, T., Seno, K., Sasaki, M., . . . Itoh, M. (2000). What are the appropriate indications for endoscopic mucosal resection for early gastric cancer? Analysis of 256 endoscopically resected lesions. Endoscopy, 32(10), 773-778. https://doi.org/10.1055/s-2000-7712
Mortensen, K., Nilsson, M., Slim, K., Schäfer, M., Mariette, C., Braga, M., . . . Lassen, K. (2014). Consensus guidelines for enhanced recovery after gastrectomy: Enhanced Recovery After Surgery (ERAS®) Society recommendations. The British journal of surgery, 101(10), 1209-1229. https://doi.org/10.1002/bjs.9582
Nakajima, T. (2002). Gastric cancer treatment guidelines in Japan. Gastric cancer : official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association, 5(1), 1-5. https://doi.org/10.1007/s101200200000
Nasjonal helseplan (2007-2010). (2006). (Særtrykk av St.prp. nr. 1 (2006-2007) kapittel 6). Oslo: Helse- og omsorgsdepartementet. Retrieved from http://www.regjeringen.no/upload/HOD/Sykehus/Nasjonal_helseplan_Sartrykk.pdf
Nasjonale faglige retningslinjer, veiledere og kvalitetsindikatorer. § 12-5 I: Lov om kommunale helse- og omsorgstjenester m.m. (helse- og omsorgstjenesteloven). LOV-2011-06-24-30. Retrieved from https://lovdata.no/lov/2011-06-24-30/§12-5
Neoadjuvant Study Using Trastuzumab or Trastuzumab With Pertuzumab in Gastric or Gastroesophageal Junction Adenocarcinoma (INNOVATION). (2015-2024). [pågående studie]. (NCT02205047). Retrieved from https://clinicaltrials.gov/ct2/show/NCT02205047
Noh, S. H., Park, S. R., Yang, H. K., Chung, H. C., Chung, I. J., Kim, S. W., . . . Bang, Y. J. (2014). Adjuvant capecitabine plus oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): 5-year follow-up of an open-label, randomised phase 3 trial. The Lancet. Oncology, 15(12), 1389-1396. https://doi.org/10.1016/s1470-2045(14)70473-5
Oda, I., Saito, D., Tada, M., Iishi, H., Tanabe, S., Oyama, T., . . . Yoshida, S. (2006). A multicenter retrospective study of endoscopic resection for early gastric cancer. Gastric cancer : official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association, 9(4), 262-270. https://doi.org/10.1007/s10120-006-0389-0
Okano, K., Maeba, T., Ishimura, K., Karasawa, Y., Goda, F., Wakabayashi, H., . . . Maeta, H. (2002). Hepatic resection for metastatic tumors from gastric cancer. Annals of surgery, 235(1), 86-91.
Pacelli, F., Cusumano, G., Rosa, F., Marrelli, D., Dicosmo, M., Cipollari, C., . . . Doglietto, G. B. (2013). Multivisceral resection for locally advanced gastric cancer: an Italian multicenter observational study. JAMA surgery, 148(4), 353-360. https://doi.org/10.1001/2013.jamasurg.309
Paoletti, X., Oba, K., Burzykowski, T., Michiels, S., Ohashi, Y., Pignon, J. P., . . . Buyse, M. (2010). Benefit of adjuvant chemotherapy for resectable gastric cancer: a meta-analysis. Jama, 303(17), 1729-1737. https://doi.org/10.1001/jama.2010.534
Park, H. S., Lee, J. M., Kim, S. H., Lee, J. Y., Yang, H.-K., Han, J. K., & Choi, B. I. (2010). Three-Dimensional MDCT for Preoperative Local Staging of Gastric Cancer Using Gas and Water Distention Methods: A Retrospective Cohort Study. American Journal of Roentgenology, 195(6), 1316-1323. https://doi.org/10.2214/AJR.10.4320
Park, S. H., Sohn, T. S., Lee, J., Lim, D. H., Hong, M. E., Kim, K. M., . . . Kang, W. K. (2015). Phase III Trial to Compare Adjuvant Chemotherapy With Capecitabine and Cisplatin Versus Concurrent Chemoradiotherapy in Gastric Cancer: Final Report of the Adjuvant Chemoradiotherapy in Stomach Tumors Trial, Including Survival and Subset Analyses. J Clin Oncol, 33(28), 3130-3136. https://doi.org/10.1200/jco.2014.58.3930
Park, S. H., Zang, D. Y., Han, B., Ji, J. H., Kim, T. G., Oh, S. Y., . . . Kang, W. K. (2019). ARTIST 2: Interim results of a phase III trial involving adjuvant chemotherapy and/or chemoradiotherapy after D2-gastrectomy in stage II/III gastric cancer (GC). J Clin Oncol, 37(Suppl 15), 4001.
Reim, D., Loos, M., Vogl, F., Novotny, A., Schuster, T., Langer, R., . . . Schuhmacher, C. (2013). Prognostic implications of the seventh edition of the international union against cancer classification for patients with gastric cancer: the Western experience of patients treated in a single-center European institution. J Clin Oncol, 31(2), 263-271. https://doi.org/10.1200/jco.2012.44.4315
Retana, A., Silverstein, T., & Wassef, W. (2011). An update in endoscopic management of gastric cancer. Current opinion in gastroenterology, 27(6), 576-582. https://doi.org/10.1097/MOG.0b013e32834b9e5e
Riihimäki, M., Hemminki, A., Sundquist, K., Sundquist, J., & Hemminki, K. (2016). Metastatic spread in patients with gastric cancer. Oncotarget, 7(32), 52307-52316. https://doi.org/10.18632/oncotarget.10740
Ronellenfitsch, U., Schwarzbach, M., Hofheinz, R., Kienle, P., Kieser, M., Slanger, T. E., & Jensen, K. (2013). Perioperative chemo(radio)therapy versus primary surgery for resectable adenocarcinoma of the stomach, gastroesophageal junction, and lower esophagus. The Cochrane database of systematic reviews, (5), CD008107. https://doi.org/10.1002/14651858.CD008107.pub2
Rudloff, U. (2018). Gastric adenocarcinoma and proximal polyposis of the stomach: diagnosis and clinical perspectives. Clin Exp Gastroenterol, 11, 447-459. https://doi.org/10.2147/ceg.S163227
Sakuramoto, S., Sasako, M., Yamaguchi, T., Kinoshita, T., Fujii, M., Nashimoto, A., . . . Arai, K. (2007). Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. The New England journal of medicine, 357(18), 1810-1820. https://doi.org/10.1056/NEJMoa072252
Sano, T., Sasako, M., Mizusawa, J., Yamamoto, S., Katai, H., Yoshikawa, T., . . . Fujitani, K. (2017). Randomized Controlled Trial to Evaluate Splenectomy in Total Gastrectomy for Proximal Gastric Carcinoma. Annals of surgery, 265(2), 277-283. https://doi.org/10.1097/sla.0000000000001814
Sasako, M., Sakuramoto, S., Katai, H., Kinoshita, T., Furukawa, H., Yamaguchi, T., . . . Ohashi, Y. (2011). Five-year outcomes of a randomized phase III trial comparing adjuvant chemotherapy with S-1 versus surgery alone in stage II or III gastric cancer. J Clin Oncol, 29(33), 4387-4393. https://doi.org/10.1200/jco.2011.36.5908
Sato, Y., Yamada, T., Yoshikawa, T., Machida, R., Mizusawa, J., Katayama, H., . . . Terashima, M. (2020). Randomized controlled Phase III trial to evaluate omentum preserving gastrectomy for patients with advanced gastric cancer (JCOG1711, ROAD-GC). Jpn J Clin Oncol, 50(11), 1321-1324. https://doi.org/10.1093/jjco/hyaa113
Shah, M. A., Janjigian, Y. Y., Stoller, R., Shibata, S., Kemeny, M., Krishnamurthi, S., . . . Kelsen, D. P. (2015). Randomized Multicenter Phase II Study of Modified Docetaxel, Cisplatin, and Fluorouracil (DCF) Versus DCF Plus Growth Factor Support in Patients With Metastatic Gastric Adenocarcinoma: A Study of the US Gastric Cancer Consortium. J Clin Oncol, 33(33), 3874-3879. https://doi.org/10.1200/jco.2015.60.7465
Shapiro, J., van Lanschot, J. J. B., Hulshof, M., van Hagen, P., van Berge Henegouwen, M. I., Wijnhoven, B. P. L., . . . van der Gaast, A. (2015). Neoadjuvant chemoradiotherapy plus surgery versus surgery alone for oesophageal or junctional cancer (CROSS): long-term results of a randomised controlled trial. The Lancet. Oncology, 16(9), 1090-1098. https://doi.org/10.1016/s1470-2045(15)00040-6
Shitara, K., Doi, T., Dvorkin, M., Mansoor, W., Arkenau, H. T., Prokharau, A., . . . Tabernero, J. (2018). Trifluridine/tipiracil versus placebo in patients with heavily pretreated metastatic gastric cancer (TAGS): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet. Oncology, 19(11), 1437-1448. https://doi.org/10.1016/s1470-2045(18)30739-3
Shitara, K., Van Cutsem, E., Bang, Y. J., Fuchs, C., Wyrwicz, L., Lee, K. W., . . . Tabernero, J. (2020). Efficacy and Safety of Pembrolizumab or Pembrolizumab Plus Chemotherapy vs Chemotherapy Alone for Patients With First-line, Advanced Gastric Cancer: The KEYNOTE-062 Phase 3 Randomized Clinical Trial. JAMA Oncol, 6(10), 1571-1580. https://doi.org/10.1001/jamaoncol.2020.3370
Shitara, K., Özgüroğlu, M., Bang, Y. J., Di Bartolomeo, M., Mandalà, M., Ryu, M. H., . . . Fuchs, C. S. (2018). Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet (London, England), 392(10142), 123-133. https://doi.org/10.1016/s0140-6736(18)31257-1
Smalley, S. R., Benedetti, J. K., Haller, D. G., Hundahl, S. A., Estes, N. C., Ajani, J. A., . . . Macdonald, J. S. (2012). Updated analysis of SWOG-directed intergroup study 0116: a phase III trial of adjuvant radiochemotherapy versus observation after curative gastric cancer resection. J Clin Oncol, 30(19), 2327-2333. https://doi.org/10.1200/jco.2011.36.7136
Smyth, E. C., Verheij, M., Allum, W., Cunningham, D., Cervantes, A., & Arnold, D. (2016). Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol, 27(suppl 5), v38-v49. https://doi.org/10.1093/annonc/mdw350
Smyth, E. C., Wotherspoon, A., Peckitt, C., Gonzalez, D., Hulkki-Wilson, S., Eltahir, Z., . . . Cunningham, D. (2017). Mismatch Repair Deficiency, Microsatellite Instability, and Survival: An Exploratory Analysis of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) Trial. JAMA Oncol, 3(9), 1197-1203. https://doi.org/10.1001/jamaoncol.2016.6762
Songun, I., Putter, H., Kranenbarg, E. M., Sasako, M., & van de Velde, C. J. (2010). Surgical treatment of gastric cancer: 15-year follow-up results of the randomised nationwide Dutch D1D2 trial. The Lancet. Oncology, 11(5), 439-449. https://doi.org/10.1016/s1470-2045(10)70070-x
Stiekema, J., Trip, A. K., Jansen, E. P., Boot, H., Cats, A., Ponz, O. B., . . . van Sandick, J. W. (2014). The prognostic significance of an R1 resection in gastric cancer patients treated with adjuvant chemoradiotherapy. Annals of surgical oncology, 21(4), 1107-1114. https://doi.org/10.1245/s10434-013-3397-4
Sumpter, K., Harper-Wynne, C., Cunningham, D., Rao, S., Tebbutt, N., Norman, A. R., . . . Oates, J. (2005). Report of two protocol planned interim analyses in a randomised multicentre phase III study comparing capecitabine with fluorouracil and oxaliplatin with cisplatin in patients with advanced oesophagogastric cancer receiving ECF. British journal of cancer, 92(11), 1976-1983. https://doi.org/10.1038/sj.bjc.6602572
Sun, J., Ilich, A. I., Kim, C. A., Chu, M. P., Wong, G. G., Ghosh, S., . . . Sawyer, M. B. (2016). Concomitant Administration of Proton Pump Inhibitors and Capecitabine is Associated With Increased Recurrence Risk in Early Stage Colorectal Cancer Patients. Clin Colorectal Cancer, 15(3), 257-263. https://doi.org/10.1016/j.clcc.2015.12.008
Syn, N. L., Wee, I., Shabbir, A., Kim, G., & So, J. B. (2019). Pouch Versus No Pouch Following Total Gastrectomy: Meta-analysis of Randomized and Non-randomized Studies. Annals of surgery, 269(6), 1041-1053. https://doi.org/10.1097/sla.0000000000003082
Søreide, J. A., Greve, O. J., Gudlaugsson, E., & Størset, S. (2016). Hepatoid adenocarcinoma of the stomach--proper identification and treatment remain a challenge. Scandinavian journal of gastroenterology, 51(6), 646-653. https://doi.org/10.3109/00365521.2015.1124286
Tanaka, N., Katai, H., Taniguchi, H., Saka, M., Morita, S., Fukagawa, T., & Gotoda, T. (2010). Trends in characteristics of surgically treated early gastric cancer patients after the introduction of gastric cancer treatment guidelines in Japan. Gastric cancer : official journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association, 13(2), 74-77. https://doi.org/10.1007/s10120-009-0536-5
Ter Veer, E., Creemers, A., de Waal, L., van Oijen, M. G. H., & van Laarhoven, H. W. M. (2018). Comparing cytotoxic backbones for first-line trastuzumab-containing regimens in human epidermal growth factor receptor 2-positive advanced oesophagogastric cancer: A meta-analysis. International journal of cancer, 143(2), 438-448. https://doi.org/10.1002/ijc.31325
Ter Veer, E., Haj Mohammad, N., van Valkenhoef, G., Ngai, L. L., Mali, R. M. A., Anderegg, M. C., . . . van Laarhoven, H. W. M. (2016). The Efficacy and Safety of First-line Chemotherapy in Advanced Esophagogastric Cancer: A Network Meta-analysis. Journal of the National Cancer Institute, 108(10). https://doi.org/10.1093/jnci/djw166
Tey, J., Back, M. F., Shakespeare, T. P., Mukherjee, R. K., Lu, J. J., Lee, K. M., . . . Zhu, M. (2007). The role of palliative radiation therapy in symptomatic locally advanced gastric cancer. International journal of radiation oncology, biology, physics, 67(2), 385-388. https://doi.org/10.1016/j.ijrobp.2006.08.070
Townsend, C. M., Beauchamp, R. D., Evers, B. M., & Mattox, K. L. (Eds.). (2017). Sabiston textbook of surgery: the biological basis of modern surgical practice (20. ed.). Philadelphia: Elsevier.
Trip, A. K., Nijkamp, J., van Tinteren, H., Cats, A., Boot, H., Jansen, E. P., & Verheij, M. (2014). IMRT limits nephrotoxicity after chemoradiotherapy for gastric cancer. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology, 112(2), 289-294. https://doi.org/10.1016/j.radonc.2014.08.039
Ueda, K., Iwahashi, M., Nakamori, M., Nakamura, M., Naka, T., Ishida, K., . . . Yamaue, H. (2009). Analysis of the prognostic factors and evaluation of surgical treatment for synchronous liver metastases from gastric cancer. Langenbeck's archives of surgery, 394(4), 647-653. https://doi.org/10.1007/s00423-008-0311-9
Van Cutsem, E., Boni, C., Tabernero, J., Massuti, B., Middleton, G., Dane, F., . . . Rougier, P. (2015). Docetaxel plus oxaliplatin with or without fluorouracil or capecitabine in metastatic or locally recurrent gastric cancer: a randomized phase II study. Ann Oncol, 26(1), 149-156. https://doi.org/10.1093/annonc/mdu496
Van Cutsem, E., Moiseyenko, V. M., Tjulandin, S., Majlis, A., Constenla, M., Boni, C., . . . Ajani, J. A. (2006). Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol, 24(31), 4991-4997. https://doi.org/10.1200/jco.2006.06.8429
van der Post, R. S., Oliveira, C., Guilford, P., & Carneiro, F. (2019). Hereditary gastric cancer: what's new? Update 2013-2018. Fam Cancer, 18(3), 363-367. https://doi.org/10.1007/s10689-019-00127-7
van der Post, R. S., Vogelaar, I. P., Carneiro, F., Guilford, P., Huntsman, D., Hoogerbrugge, N., . . . Fitzgerald, R. C. (2015). Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. Journal of medical genetics, 52(6), 361-374. https://doi.org/10.1136/jmedgenet-2015-103094
van Hagen, P., Hulshof, M. C., van Lanschot, J. J., Steyerberg, E. W., van Berge Henegouwen, M. I., Wijnhoven, B. P., . . . van der Gaast, A. (2012). Preoperative chemoradiotherapy for esophageal or junctional cancer. The New England journal of medicine, 366(22), 2074-2084. https://doi.org/10.1056/NEJMoa1112088
Viste, A., Eide, G. E., Halvorsen, K., Maartmann-Moe, H., & Søreide, O. (1986). The prognostic value of Lauren's histopathological classification system and ABO blood groups in patients with stomach carcinoma. European journal of surgical oncology : the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology, 12(2), 135-141.
Viste, A., Haùgstvedt, T., Eide, G. E., & Soreide, O. (1988). Postoperative complications and mortality after surgery for gastric cancer. Annals of surgery, 207(1), 7-13.
Waddell, T., Verheij, M., Allum, W., Cunningham, D., Cervantes, A., & Arnold, D. (2014). Gastric cancer: ESMO-ESSO-ESTRO clinical practice guidelines for diagnosis, treatment and follow-up. European journal of surgical oncology : the journal of the European Society of Surgical Oncology and the British Association of Surgical Oncology, 40(5), 584-591. https://doi.org/10.1016/j.ejso.2013.09.020
Wagner, A. D., Unverzagt, S., Grothe, W., Kleber, G., Grothey, A., Haerting, J., & Fleig, W. E. (2010). Chemotherapy for advanced gastric cancer. The Cochrane database of systematic reviews, (3), CD004064. https://doi.org/10.1002/14651858.CD004064.pub3
Wasmuth, H. H., Thorsen, G., Nordgård, K., & Gjesdahl, C. (1997). Ventrikkelkreft. Et tiårsmateriale med vekt på tidligkreft. Tidsskrift for den Norske laegeforening : tidsskrift for praktisk medicin, ny raekke, 117(7), 931-934.
Wei, W. Z., Yu, J. P., Li, J., Liu, C. S., & Zheng, X. H. (2005). Evaluation of contrast-enhanced helical hydro-CT in staging gastric cancer. World J Gastroenterol, 11(29), 4592-4595.
WHO Classification of Tumours Editorial Board (Ed.). (2019). Digestive System Tumours: WHO Classification of Tumours (5. ed.). Geneva: World Health Organization.
Wilke, H., Muro, K., Van Cutsem, E., Oh, S. C., Bodoky, G., Shimada, Y., . . . Ohtsu, A. (2014). Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. The Lancet. Oncology, 15(11), 1224-1235. https://doi.org/10.1016/s1470-2045(14)70420-6
Yasuda, K., Shiraishi, N., Suematsu, T., Yamaguchi, K., Adachi, Y., & Kitano, S. (1999). Rate of detection of lymph node metastasis is correlated with the depth of submucosal invasion in early stage gastric carcinoma. Cancer, 85(10), 2119-2123.
Ychou, M., Boige, V., Pignon, J. P., Conroy, T., Bouche, O., Lebreton, G., . . . Rougier, P. (2011). Perioperative chemotherapy compared with surgery alone for resectable gastroesophageal adenocarcinoma: an FNCLCC and FFCD multicenter phase III trial. J Clin Oncol, 29(13), 1715-1721. https://doi.org/10.1200/jco.2010.33.0597
Yu, C., Yu, R., Zhu, W., Song, Y., & Li, T. (2012). Intensity-modulated radiotherapy combined with chemotherapy for the treatment of gastric cancer patients after standard D1/D2 surgery. Journal of cancer research and clinical oncology, 138(2), 255-259. https://doi.org/10.1007/s00432-011-1085-y
Yu, W., Choi, G. S., & Chung, H. Y. (2006). Randomized clinical trial of splenectomy versus splenic preservation in patients with proximal gastric