









Veileder for diagnostikk og behandling av amyloidose
Her kan du laste ned en utskriftsvennlig PDF-utgave av veilederen.
Forside
Tale Norbye Wien, Ann Kristin Kvam, Maria Stensland, Kristin Ørstavik, Einar Gude, Ketil Riddervold Heimdal, Fredrik Hellem Schjesvold, Anders Hodt , Tore Bach-Gansmo, Helga Gudmundsdottir, Svein Oskar Frigstad, Guri Hagberg, Per Selnes og Melinda Raki
2. reviderte utgave av veilederen er ferdigstilt desember 2021.
Forord
Forord til 2. utgave 2021
Med de siste årenes framskritt på feltet er veilederen fra 2016 nå moden for oppdatering. Målet med den nye utgaven av veilederen er som sist å gi en lett tilgjengelig oppdatering om diagnostikk og behandling. Veilederen er oppdatert med nytt kapittel om organmanifestasjoner og nye tabeller og flytdiagrammer for utredning. Siden 2016 har en nukleærmedisinsk undersøkelse (amyloidoseskjelettscintigrafi) fått en sentral plass i utredning av mistenkt hjerteamyloidose. For typing brukes i økende grad lasermikrodisseksjon med massespektrometri som supplement til immunhistokjemi. Behandlingsmuligheter for amyloidose forbedres stadig, og veilederen vil her presentere prinsipper for behandling. Veilederen kan ikke holdes à jour fortløpende etter publiseringstidspunkt og leseren bes konsultere oppdatert litteratur. For AL-amyloidose vises spesielt til Handlingsprogram for AL-amyloidose og til Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligne blodsykdommer, begge elektronisk tilgjengelig på nettsidene til Norsk selskap for hematologi (1, 2).
Oppdateringen er som sist utarbeidet på det forfatterne mener er et fritt faglig grunnlag uten ekstern finansiering fra industrien eller andre kilder. Flere medforfattere fra flere spesialiteter har bidratt til den nye utgaven, viser til forfatterliste på side 6. Forfatterne er selv ansvarlige for innholdet slik det foreligger per desember 2021. Den elektroniske versjonen er tilgjengelig på nettsidene til henholdsvis Helsebiblioteket, Den norske patologforening, Norsk selskap for hematologi og Norsk nyremedisinsk forening.
Forord til 1. utgave 2016
Hensikten med denne veilederen er å gi norske leger og andre interesserte en lett tilgjengelig oppdatering om diagnostikk og behandling av amyloidose som kan være nyttig i praktisk klinisk arbeid. Den klassiske gamle inndeling i primær og sekundær amyloidose er for lengst utdatert, og er erstattet av en flora av amyloidosetyper. Tidlig og riktig diagnose er viktig da behandlingsmulighetene både økes og endres pga. teknologiske fremskritt. Dette gir berettiget håp om bedring av livskvalitet og leveutsikter.
Det kliniske bildet er mangfoldig og både allmennleger, ulike spesialister og grenspesialister vil møte pasienter med kliniske symptomer og tegn der muligheten for amyloidose bør overveies.
Forfatterne er en selvoppnevnt faggruppe som med ulike innfallsvinkler i lang tid har interessert seg for amyloidose. Formelt har de bakgrunn i ulike fagfelt: Biokjemi, patologi, generell indremedisin, gastroenterologi, revmatologi, nyresykdommer og blodsykdommer. De har forskningsbakgrunn og/eller relativt omfattende klinisk erfaring med pasientgruppen. Gruppen har i forbindelse med veilederen knyttet til seg kardiolog og medisinsk genetiker som har bidratt på sine felt og gruppen har dessuten innhentet synspunkter og råd fra gastroenterolog og hepatolog Kristian Bjøro, som takkes for dette.
Forfatterne er selv ansvarlige for innholdet slik det foreligger per august 2016. Publikasjonen er utarbeidet på det forfatterne mener er et fritt faglig grunnlag uten ekstern finansiering fra industrien eller andre kilder. Bidrag til trykking og distribusjon av papirversjonen er gitt fra legemiddelfirmaet Celgene. Den elektroniske versjonen er tilgjengelig på nettsidene til Den norske patologforening, Norsk selskap for hematologi og Norsk nyremedisinsk forening.
Forfatterne
Tale Norbye Wien. Dr. med. Spesialist i indremedisin og nyresykdommer. Overlege ved medisinsk avdeling, Bærum sykehus, Vestre Viken.
Ann Kristin Kvam. PhD. Spesialist i indremedisin og blodsykdommer. Overlege ved avdeling for blodsykdommer, Oslo universitetssykehus.
Fredrik Hellem Schjesvold. PhD. Spesialist i indremedisin. Overlege/forsker ved avdeling for blodsykdommer, Oslo Universitetssykehus.
Einar Gude. Dr. med. Spesialist i indremedisin og hjertesykdommer. Seksjonsoverlege ved kardiologisk avdeling, Oslo universitetssykehus.
Helga Gudmundsdottir. PhD. Spesialist i indremedisin og nyresykdommer. Overlege ved Nyremedisinsk avdeling, Oslo universitetssykehus.
Svein Oskar Frigstad. Spesialist i indremedisin og fordøyelsessykdommer. Overlege ved medisinsk avdeling, Bærum sykehus, Vestre Viken.
Guri Hagberg. PhD. Spesialist i indremedisin og geriatri. Overlege ved seksjon for hjerneslag, Oslo universitetssykehus. Seniorforsker tilknyttet Forskningsavdelingen Bærum Sykehus, Vestre Viken.
Per Selnes. PhD. Spesialist i nevrologi. Overlege ved nevrologisk avdeling, Akershus Universitetssykehus.
Kristin Ørstavik. Dr.med. Spesialist i nevrologi og klinisk nevrofysiologi. Seksjonsleder, seksjon for sjeldne nevromuskulære tilstander, Nevrologisk avdeling, Oslo Universitetssykehus.
Ketil Riddervold Heimdal. Dr. med. Spesialist i medisinsk genetikk. Overlege ved avdeling for medisinsk genetikk, Oslo universitetssykehus.
Tore Bach-Gansmo. Dr. med. spesialist i nukleærmedisin, Overlege ved PET og Nukleærmedisin, Universitetssykehuset Nord-Norge, Tromsø.
Anders Hodt. PhD. Spesialist i nukleærmedisin. Overlege ved avdeling for nukleærmedisin, Oslo universitetssykehus.
Maria Stensland. PhD i proteomikk/immunologi. Avdelingsingeniør ved Immunologisk Institutt/Kjernefasilitet for proteomikk, Oslo Universitetssykehus.
Melinda Raki. PhD. Spesialist i patologi. Overlege ved avdeling for patologi, Oslo universitetssykehus.
Forfattere 1. utgave 2016: Tale Norbye Wien, Fredrik Schjesvold, Gunnar Husby, Lorentz Brinch, Tobias Gedde-Dahl, Knut Sletten, Erik Heyerdahl Strøm, Gustavo de Souza, Maria Stensland, Ketil Riddervold Heimdal, Einar Gude
Potensielle interessekonflikter
Tale Norbye Wien og Einar Gude har mottatt honorar for foredrag og bidrag i ekspertpanel fra Pfizer, Alnylam og Janssen-Cilag. Ann Kristin Kvam har mottatt honorar for foredrag fra Janssen og BMS. Fredrik Schjesvold har mottatt honorar for foredrag og bidrag i ekspertpanel fra Amgen, BMS, Takeda, Abbvie, Janssen, Novartis, SkyliteDx, Oncopeptides, Sanofi, Pfizer, Daiki-Sankyo, GSK, Celgene. Øvrige forfattere oppgir ingen potensielle interessekonflikter.
Forkortelser
AA = Amyloid A
Aβ = Amyloid beta
Aβ2M = Amyloid beta 2-mikroglobulin
AD = Alzheimers demens
AEF = Amyoid enhancing factor
AFib = Fibrinogenamyloid
AKR = Albumin/kreatinin ratio
AL = Lettkjede-amyloid
ALECT2 = Leukocytt kjemotaktisk faktor 2-amyloid
ATTR = Transtyretin-amyloid
ATTRv = ATTR med v for variant, dvs. arvelig form
ATTRwt = ATTR med wt for villtype, dvs. ikke arvelig form
CAA = Cerebral amyloid angiopati
CRP = C-reaktivt protein
CSF = Cerebrospinalvæske
CTS = Karpaltunnelsyndrom
DPD = 99mTc-DPD (3,3-Difosfono-1,2-propan-dikarboksylsyre)
EMA = European Medical Agency
EMG = Elektromyografi
FDA = US Food and Drug Administration
FDG-PET = 18FFluorodeksyglukose positronemisjonstomografi
FLC = Frie lette immunglobulinkjeder
dFLC = Differanse mellom involverte og ikke-involverte lettkjeder
FMF = Familiær middelhavsfeber
GFR = Glomerulær filtrasjonsrate
HCM = Hypertrofisk kardiomyopati
HFpEF = Hjertesvikt med bevart ejeksjonsfraksjon
HMDP = 99mTc-HMDP (hydroxymethylenediphosphonate)
HMAS = Høydose kjemoterapi med autolog stamcellestøtte
ICD = Implanterbar hjertestarter
INR = International Normalized Ratio
LMD-MS = Laserdisseksjon med påfølgende massespektrometri
MGUS = Monoklonal gammopati av usikker klinisk signifikans
NTproBNP = N-terminalt pro-B-type natriuretisk peptid
PET = Positronemisjonstomografi
PKR = totalprotein/kreatinin ratio
PYP = 99mTc-PYP, pyrofosfat
SAA = Serumprotein amyloid A
SAP = Serum amyloid P komponent
SPECT = Single Photon Emission Computed Tomography
TnT = Troponin T
VGPR = Very good partial response
VVH = Venstre ventrikkel hypertrofi
1. Introduksjon
1.1 Mål
Målet med denne veilederen er å formidle oppdatert kunnskap om diagnostikk og behandling av amyloidose.
Sentralt i dette er å:
- Mistenke amyloidose og sette i gang utredning av om det foreligger en amyloidose
- Påvise amyloidose i vevsprøve
- Vurdere om det er systemisk eller lokalisert affeksjon
- Avgjøre hvilken type amyloidose det dreier seg om
- Sette inn behandling (når slik finnes)
Denne veilederen er en oppdatert versjon av veilederen som ble publisert i 2016, og var utarbeidet av Arbeidsgruppe for amyloidose. Arbeidsgruppen er selvoppnevnt og etablerte seg i 2010 med mål å forbedre diagnostikk, typing og behandling av amyloidose i Norge. Vi har sett behov for vårt arbeid begrunnet i internasjonal rapportering av feildiagnoser (3, 4), forsinkelser i diagnostikk (5, 6) og nasjonal rapportering av underdiagnostikk (7). Arbeidsgruppen har organisert et tverrfaglig forum for diagnostikk, typing og behandling av amyloidose med representanter fra hematologi, kardiologi, nefrologi, nevrologi, nukleærmedisin, bioteknologi, medisinsk genetikk, immunologi, proteinsekvensering og patologi (8).
1.2 Grunnlaget for anbefalingene
Anbefalingene er basert på tilgjengelige studier og litteratur om amyloidose gjennom ikkesystematiske søk i PubMed samt arbeidsgruppens egne erfaringer fra arbeid med utredning av og behandling av amyloidose. Anbefalingene gjelder for tidspunktet for ferdigstillelse.
1.3 Definisjon og karakteristika
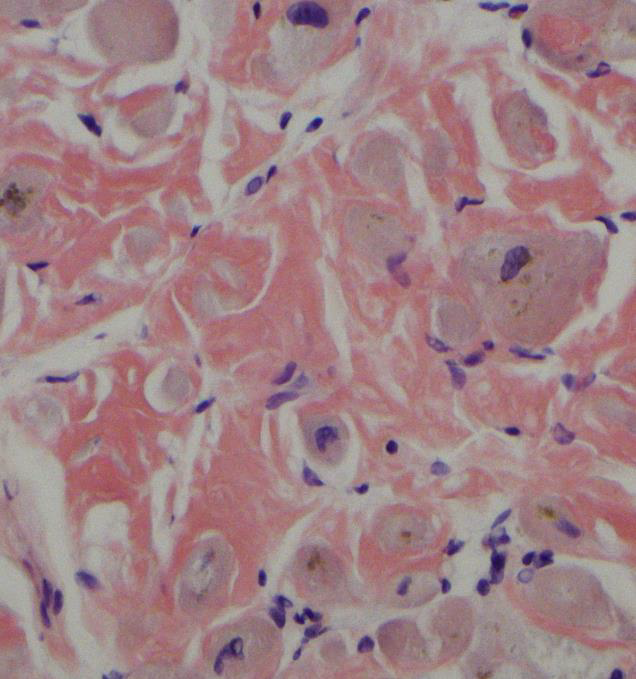
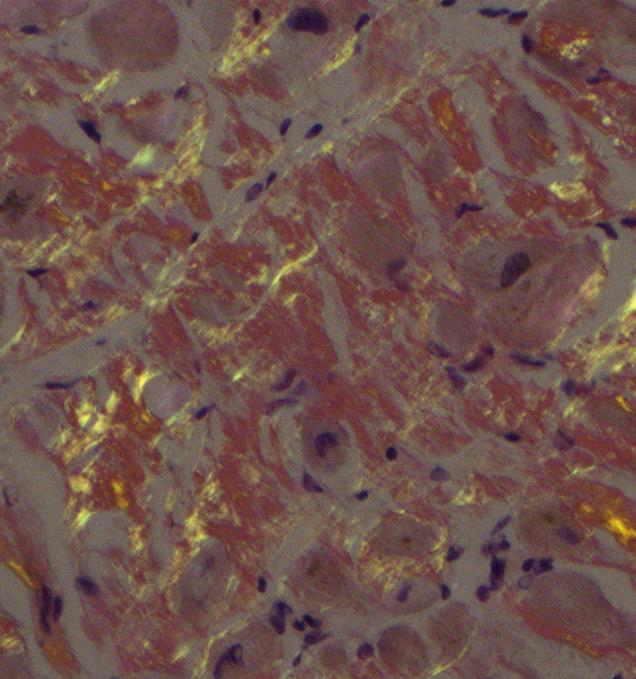
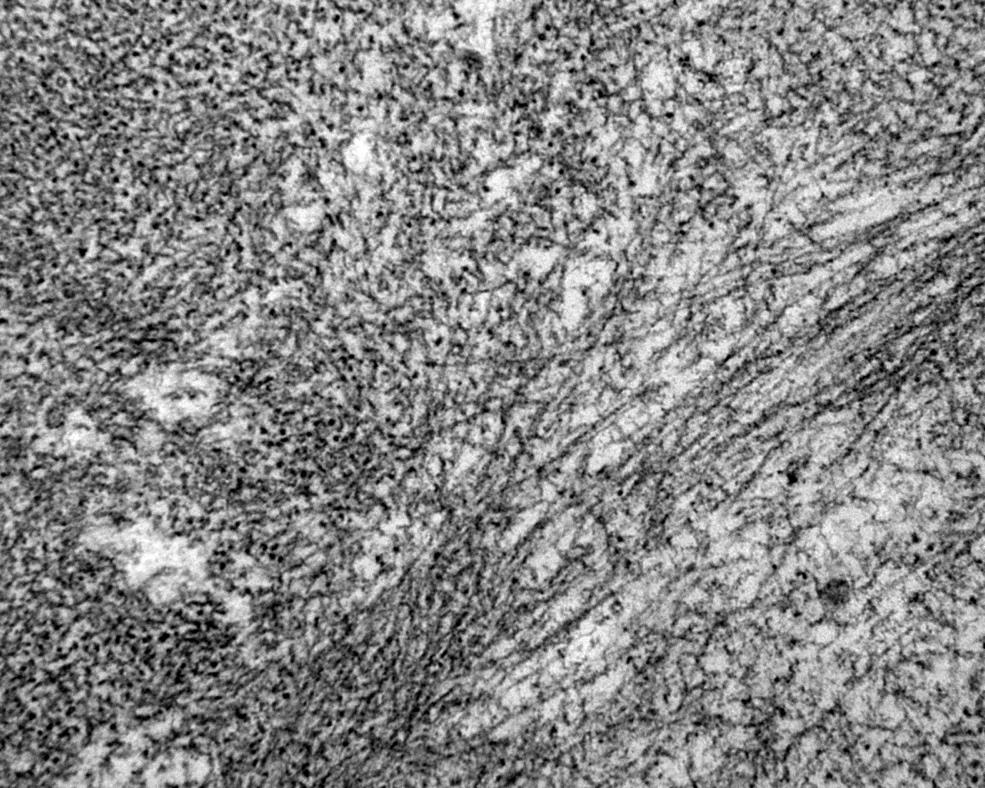
Amyloidose er en gruppe sykdommer som kjennetegnes ved avleiring av polypeptidmateriale ekstracellulært i organer og vev. Dette materialet kalles amyloid, og betegnelsen stammer fra gresk amylon, som betyr stivelse, og kommer av at avleiringene har egenskaper som minner om stivelse i histologiske snitt. Hovedkomponenten i amyloid er amyloide fibriller, som består i hovedsak av et protein, deler av et protein eller bare et peptid som danner fibriller. Disse fibrillene er endestadiet i en sekvensiell forandring i polypeptidets folding som resulterer i en form som er karakteristisk for amyloidose. Det amyloide fibrilleproteinet varierer med typen av amyloidose, men de ulike fibrilleproteinene har alle en karakteristisk og felles ultrastruktur (9). Fibrillene kan påvises ved elektronmikroskopi (Figur 1) og har helt spesielle egenskaper; de binder blant annet fargestoffet kongorødt som ved polarisert lys gir en gul/oransje-grønn dobbeltbrytning (Figur 2). Farging med kongorødt benyttes rutinemessig for å påvise amyloid i vevsprøver.
Dannelsen og avleiringen av amyloid forårsaker sviktende organfunksjon ved å fortrenge normale vevskomponenter og vevsfunksjoner. Avleiringen er en dynamisk prosess som potensielt kan være reversibel, men gir ofte irreversibel organskade. Amyloid kan avleires i alle kroppens organer, enten i ett organ (lokalisert amyloid) eller i flere organsystemer (systemisk amyloidose).
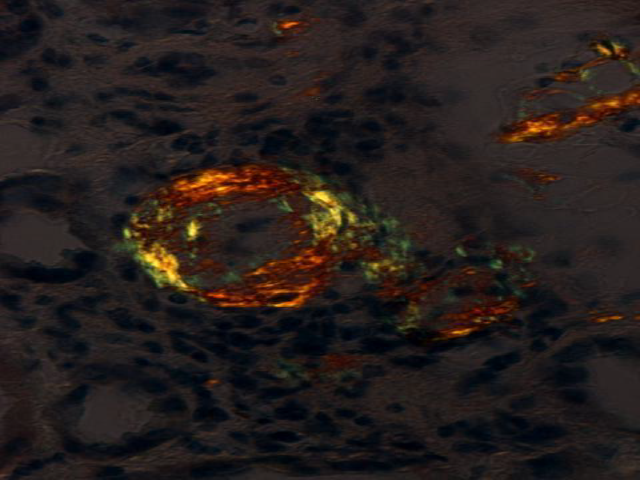
1.4 Klassifisering og typing
Amyloidosene klassifiseres i dag ut fra fibrilleproteinet, det protein som danner de amyloide fibrillene. Det er, med sjeldne unntak, bare ett fibrilleprotein til stede i det deponerte amyloidet i hvert enkelt tilfelle av amyloidose (10, 11). Amyloidtypen forkortes med stor A for amyloid, med påfølgende forkortelse for det angjeldende proteinet, f.eks. AA for amyloid dannet fra proteinet serum amyloid A (SAA), ATTR for amyloid dannet fra transtyretin (TTR), og AL for amyloid dannet fra lette immunglobulinkjeder. Videre spesifisering kan gis etter proteinnavnet, f.eks. ATTRwt eller ATTRv (wt = villtype og v = variant). Videre inndeler man vanligvis amyloidosene etter deres deponeringsmønster i systemiske og lokaliserte former og ut fra arvelige og ervervede former.
I dette dokumentet brukes ervervet amyloidose om amyloidoser som forårsakes av normale proteiner (inkludert lette kjeder) som danner amyloid, mens de arvelige (hereditære) er forårsaket av varianter, dvs. medfødte muterte proteiner. De arvelige amyloidosene vil opptre som en monogen, oftest autosomalt dominant, sykdom i familier. Mutasjonene ved arvelige amyloidoser viser høy, men ikke fullstendig, penetrans (penetrans = risiko for sykdom gitt at personen er mutasjonsbærer). Klinisk sykdom vil derfor ofte kunne hoppe over et slektsledd. Hvert av fibrilleproteinene er assosiert med en eller flere kliniske uttrykksformer (Tabell 1) (12).
Per i dag kjenner man 36 ulike proteiner som kan danne amyloide fibriller i mennesket (Tabell 1)(12). Av disse opptrer 18 som systemisk amyloidose og 22 som lokalisert amyloidose, og for noen proteiner finne både systemiske og lokaliserte former. Listen over amyloiddannende proteiner oppdateres av Nomenklaturkomiteen i International Society of Amyloidosis ca. annet hvert år. Per i dag er det flere kandidatproteiner som er publisert og danner amyloid som venter på godkjenning som amyloide proteiner av Nomenklaturkomiteen, bl.a. paratyroidhormon (PTH) (13) og glukagon (14).
Identifisering av det amyloide fibrilleproteinet, dvs. typing av amyloidosen, samt å fastslå om det er systemisk eller lokalisert sykdom er helt avgjørende for riktig behandling og kan ha stor betydning for sykelighet og overlevelse. Amyloidtyping vil bli nærmere gjennomgått i kapittel 3 om diagnostikk. Videre må man forsøke å avgjøre om det dreier seg om en arvelig amyloidose. Flere fibrilleproteiner kan danne amyloid både med den muterte og den native formen som utgangsmateriale, og gi dermed opphav til både arvelig og ikke-arvelig amyloidose.
Betegnelsene primær og sekundær amyloidose anbefales ikke lenger brukt da amyloidose skal benevnes etter det amyloide fibrilleproteinet.
Flere sykdommer hvor amyloide fibriller er en del av sykdomsprosessen blir per i dag vanligvis ikke omtalt som amyloidoser, selv om fibrilleproteinet regnes med blant amyloidosene (12). Dette gjelder Alzheimers sykdom (amyloid protein: Aβ) (15), Parkinsons sykdom (amyloid protein: AαSyn) (16) og type 2 diabetes (amyloid protein: AIAPP) (17, 18).
1.5 Klinisk uttrykksform
Alle kroppens organer kan affiseres av en eller flere typer amyloidose, og symptomene på amyloidose vil følgelig avhenge av hvilke organer som rammes i det enkelte tilfellet (se kapittel 4). Typiske, men uspesifikke, manifestasjoner ved systemisk amyloidose er bl.a. vekttap, slapphet, tretthet, proteinuri med eller uten nyresvikt, restriktiv kardiomyopati, gastrointestinale og respiratoriske symptomer, hepatomegali, hudaffeksjon evt. med purpura, polynevropati og karpaltunnelsyndrom. Affeksjon av nyre og hjerte er det som vanligvis gir symptomer ved de hyppigst forekommende systemiske amyloidosene. Symptomer ved ulike amyloidoser vil bli nærmere gjennomgått i kapittel om organmanifestasjoner og undergrupper av amyloidose (kapittel 4 og 5).
Tabell 1. Amyloide fibrilleproteiner og deres forløperprotein (prekursor) i mennesket. Modifisert etter Benson og medarbeidere 2020 (12).
Amyloidose | Fibrilledannende protein | S/L | E/H | Målorganer |
AL | Immunglobulin lette kjeder | S, L | E | Alle organer, vanligvis unntatt SNS |
AH | Immunglobulin tunge kjeder | S, L | E, H | Alle organer unntatt SNS |
AA | (Apo) Serum amyloid A | S | E | Alle organer unntatt SNS |
Aβ2M | β2-microglobulin, villtype, variant | L, S | E, H | Muskel/skjelettsystemet, ANS |
ATTRwt ATTRv | Transtyretin, villtype Transtyretin, varianter | S S | E H | Hjerte PNS, ANS, hjerte, øye, leptomeninger |
AApoAI | Apolipoprotein A I, varianter | S | H | Hjerte, lever, nyre, PNS, testis, larynx, hud |
AApoAII | Apolipoprotein A II, varianter | S | H | Nyre |
AApoAIV | Apolipoprotein A IV, villtype | S | E | Nyremarg og systemisk |
AApoCII | Apolipoprotein C II, varianter | S | H | Nyre |
AApoCIII | Apolipoprotein C III, varianter | S | H | Nyre |
AGel | Gelsolin, varianter | S | H | PNS, kornea, nyre |
ALys | Lysozym, varianter | S | H | Nyre |
ALECT2 | Leukocytt kjemotaktisk faktor 2 | S | E | Nyre (primært) |
AFib | Fibrinogen α, varianter | S | H | Nyre (primært) |
ACys | Cystatin C, varianter | S | H | SNS, PNS, hud |
ABri | ABriPP, varianter | S | H | SNS |
ADan | ADanPP, varianter | S | H | SNS |
Aβ Aβ | Aβ protein prekursor, villtype Aβ protein prekursor, variant | L L | E H | SNS SNS |
AαSyn | α-Synuclein | L | E | SNS |
ATau | Tau | L | E | SNS |
APrP | Prion protein, villtype, varianter | L | E, H | SNS (bl.a. Creutzfeldt Jacobs sykdom), PNS |
ACal | (Pro)calcitonin | L,S | E | Tyreoideatumores (C-celler), nyre |
AIAPP | Islet amyloid polypeptid (Amylin) | L | E | Langerhanske øyer, insulinom |
AANF | Atrial natriuretisk faktor | L | E | Hjertets atrier, aldersrelatert |
APro | Prolaktin | L | E | Prolaktinomer i hypofysen, aldersrelatert |
AIns | Insulin | L | E | Iatrogen, lokal injeksjon |
ASPC | Lungesurfaktant protein C | L | E | Lunge |
ACor | Korneodesmosin | L | E | Hud: Liktorner og hårfollikler |
AMed | Laktadherin | L | E | Aorta-media, aldersrelatert |
AKer | Keratoepitelin | L | E, H | Hud, hornhinne, arvelig |
ALac | Laktoferrin | L | E | Hornhinne |
AOAAP | Odontogent ameloblastassosiert protein | L | E | Odontogene tumores |
ASem1 | Semenogelin 1 | L | E | Vesikula seminalis |
AEnf | Enfurvitide | L | E | Iatrogen, lokal injeksjon |
ACatK | Cathepsin K | L | E | Tumorassosiert |
AEFEMP1 | EGF-containing fibulin-like extracellular matrix protein 1 | L | E | Portvener, aldersassosiert |
Forkortelser: SNS: sentralnervesystemet, PNS: perifere nervesystem, ANS: autonome nervesystem. S: Systemisk, L: Lokalisert, E: Ervervet, H: Hereditær (arvelig)
1.6 Epidemiologi
Så vel norske som internasjonale data for insidens og prevalens av amyloidose er mangelfulle. Populasjonsbaserte studier har vært sparsomme, da amyloidose har vært regnet som en sjelden sykdom og pasientene ofte har blitt fulgt eller diagnostisert på tertiærsentra. Ut fra data fra utenlandske autopsistudier og dødsårsaksregistre er det imidlertid sannsynlig at amyloidose er underdiagnostisert også i Norge og at et betydelig antall pasienter ikke får adekvat diagnostikk og behandling (19). Økende insidens av systemisk amyloidose internasjonalt de siste årene antas å skyldes økende oppmerksomhet om sykdommen, og spesielt for transtyretinamyloidose (ATTR) i hjertet, innføring av et non-invasivt diagnostisk alternativ til biopsi (19, 20).
Insidenstall av systemisk amyloidose på 0,8/100 000 personer/år for perioden 2001-2008 har vært vist fra både Sverige og i Storbritannia i populasjonsbaserte studier basert på henholdsvis diagnoseregisteret for sykehusinnleggelser og polikliniske konsultasjoner ved svenske sykehus (21), og på henvisninger til det britiske nasjonale amyloidosesenteret samt det britiske nasjonale dødsårsaksregisteret (22). I begge disse populasjonene er insidensen høyest i aldersgruppen 60-80 år. Siden dataene fra tidlig 2000-tall ble publisert har det britiske nasjonale amyloidosesenteret senere rapportert en økning på 670 % i nye tilfeller fra perioden 1987-1999 til 2010-2019 (20). En flerdobling av antall påviste tilfeller av systemisk amyloidose over den siste 30-års perioden er også vist fra amerikanske tertiærsentre (23).
De hyppigst forekommende systemiske amyloidoser er ATTR, AL og AA. Prognosen av ubehandlet systemisk amyloidose er dokumentert i eldre studier; med median overlevelse 6-12 mnd. ved AL, 3-4 år ved AA amyloidose og ca. 5 år ved ATTRwt (21, 22, 24).
Insidensen av AL-amyloidose antas å ha liten geografisk variasjon, og har vært estimert til 0,3- 1,6/100 000 personer/år (22, 25, 26). Data fra en amerikansk sykehusdatabase som dekker ca. 70 mill. innbyggere i perioden 2007-2015 viste stabil insidens av AL på 1-1,4/100 000 personer/år, og en økende prevalens fra 2007 til 2015 på 1,6 til 4,1/100 000 personer/år (27).
Insidensen av AA-amyloidose varierer med forekomsten av underliggende inflammatoriske sykdommer og behandlingen av disse, og har derfor hatt betydelig geografisk variasjon. Prevalens ved kroniske inflammatoriske sykdommer har vært estimert til 5-10 %, eller høyere hvis asymptomatiske individer også medregnes (28). Ettersom inflammatorisk revmatisk sykdom har vært hovedårsak til AA-amyloidose i Norge og vestlige land, har insidensen av AA-amyloidose gått ned når denne gruppen de senere år har fått mer effektiv antiinflammatorisk behandling mot sin grunnsykdom. Kroniske suppurative infeksjoner som følge av injisering av rusmidler er en økende årsak til AA-amyloidose, også i Norge (29).
Ervervet transtyretinamyloidose (ATTRwt), tidligere kalt senil systemisk amyloidose, er i autopsistudier påvist hos ca. 25 % av befolkningen over 80 års alder, men gir langt sjeldnere klinisk sykdom (30, 31). Hos pasienter som utredes for TAVI (trans aortal valve insertion) er det vist at ca. 16 % har amyloidose ved amyloidose-skjelettscintigrafi (32). Arvelige amyloidoser har vært lite undersøkt i Norge, men enkelttilfeller av ulike typer arvelig amyloidose er kjent. Det er hittil dog ikke påvist arvelig ATTR-amyloidose hos etnisk norske og noen «norsk» founder-mutasjon i TTR-genet er ikke funnet.
Av norske data har vi oversikt over forekomst av nyreamyloidose i populasjonen som er etablert i nyreerstattende behandling (dvs. dialyse eller nyretransplantasjon); disse registreres i Norsk Nyreregister. I perioden 1980-2020 var det 544 pasienter som hadde amyloidose som årsak til behov for nyreerstattende behandling. Gjennomsnittet var på 13 nye pasienter årlig, med spredning på 6- 22 nye pasienter per år (33). I dette registeret er type amyloidose ikke angitt.
Når det gjelder lokaliserte amyloidoser er tallene også mangelfulle. Nodulære deposisjoner ved lokalisert amyloidose kan etterligne malignitet, f.eks. i lunge og urinblære.
Lokalisert amyloid kan sees ved de relativt hyppig forekommende sykdommene Alzheimers sykdom (AD), type 2 diabetes og cerebral amyloid angiopati (CAA). AD utgjør om lag 50-70 % av alle demenstilfeller, med økende prevalens grunnet en aldrende befolkning. Kortikale ekstracellulære amyloide plakk og intracellulære nevrofibrillære floker er de grunnleggende nevropatologiske forandringene ved Alzheimers sykdom. Rollen til amyloide fibriller i patogenesen til Alzheimers sykdom har tidligere vært omdiskutert, men det er i dag enighet om at amyloide plakk er en essensiell og tidlig faktor i utviklingen av Alzheimers sykdom (15). Ved CAA akkumuleres amyloid i hjernens blodårer, kortikale og leptomeningiale arterioler, kapillærer og vener. CAA er vanlig med en estimert prevalens på 20-40 % hos kognitivt friske 80-90 åringer, mot 50-60 % av eldre med demens (34).
Ved avdeling for patologi ved OUS-HF stilles en amyloiddiagnose ca. 100-120 ganger per år, hyppigst i biopsier fra nyre etterfulgt av hjerte, benmarg, gastrointestinaltraktus, hud, lunge og fettvev.
1.7 Nytt siden 2016-utgaven og framskritt på feltet
Den nye utgaven av veilederen er oppdatert og utvidet siden forrige utgave fra 2016. Flere nye fibrilleproteiner som danner amyloid i mennesket er karakterisert (12) og disse er inkludert i Tabell 1. Typing av amyloidet basert på immunhistokjemi gjøres ved flere norske patologilaboratorier, stort sett begrenset til AA, AL og evt. ATTR. Avdeling for patologi ved Oslo universitetssykehus tar nå imot vevsprøver fra hele landet for utvidet typing med immunhistokjemi og lasermikrodisseksjon med påfølgende massespektrometrisk typing.
I kapittel 3 om diagnostikk er omtalen av nukleærmedisinsk påvisning av amyloid i hjertet med amyloidose-skjelettscintigrafi utvidet. Denne metoden er de siste årene godt etablert i diagnostikken av ATTR-hjerteamyloidose og er nå tilgjengelig i alle helseregioner for utredning av mistenkt hjerteamyloidose. Den scintigrafiske undersøkelsen kan redusere behov for biopsi, når ALamyloidose er utelukket (35). Avsnittene om utredning av organutbredelse er utvidet, bl.a. med nye avsnitt om undersøker av mage-tarmtraktus, nevrofysiologiske undersøkelser og flytskjema for utredning av hjerteamyloidose.
Omtale av ulike organmanifestasjoner av amyloidose er utvidet og blir presentert i nytt kapittel 4. Hovedfokus er på organmanifestasjoner av systemiske amyloidoser. I kapittel 5 om de ulike amyloidosene er seksjonen om AA-amyloidose utvidet med flytskjema for utredning av bakenforliggende årsaker til AA-amyloidose. Seksjonen om lokaliserte amyloidoser er utvidet med mer om amyloid i sentralnervesystemet.
I kapittel 6 om behandling gjennomgås organstøttende behandling ved systemisk amyloidose samt prinsipper for amyloidrettet terapi som er tilgjengelig på tidspunkt for ferdigstilling av denne utgaven desember 2021. Ulike angrepspunkter for behandling vil bli kort presentert inklusiv antistoffbaserte, genteknologisk baserte og konformasjonsstabiliserende terapi. Spesielt for AL- og ATTR-amyloidose har det vært en rivende utvikling av amyloidrettet terapi de siste årene, og denne utviklingen forventes å fortsette. Veilederen tar ikke mål av seg til å kunneholdes à jour fortløpende og leseren bes konsultere oppdatert litteratur for basalforskning og behandling.
2. Patogenese
Hvilke mekanismer som er involvert i dannelsen av amyloide fibriller er ikke kjent i detalj, men in vitro studier har vist at fibrilledannelsen foregår i flere trinn. Isolerte amyloide fibriller har helt spesielle egenskaper som er forskjellige fra andre typer fibriller. Amyloide fibriller er i hovedregel ikke forgrenede. Fibrillenes lengde kan variere; mens fibrillediameteren (tykkelsen) er 8-10 nm (se Figur 1 ovenfor).
Elektronmikroskopi og røntgenkrystallografistudier av amyloide fibriller viser at de har en karakteristisk beta-plate struktur som benevnes som «cross-beta» (36).
Følgende faktorer bidrar i dannelsen av amyloide fibriller:
- Strukturelle egenskaper ved de amyloide fibrilleproteinene: Bestemte punktmutasjoner i genet som endrer proteinets tredimensjonale struktur, forandringer i det proteolytiske miljøet som resulterer i en annen type degradering, og aldersrelaterte forandringer er kjent fra flere typer av amyloidose. De fleste arvelige amyloidoser er resultat av punktmutasjoner i genet som koder for de respektive fibrilleproteinene. Feilfolding eller proteolytisk spalting av proteinet kan eksponere ellers skjulte regioner av polypeptidkjeden. Dette disponerer for interaksjon med andre molekyler og aggregering. En teori er at cellenes kvalitetskontrollsystem som hindrer sekresjon av feilfoldede proteiner, svekkes med alderen og fører til utskillelse av disse proteinene med påfølgende amyloiddannelse (37).
- Overskudd av forløper-(prekursor-) proteinet til det amyloide fibrilleproteinet: Enten som følge av økt produksjon eller nedsatt nedbrytning, evt. en kombinasjon av disse. Fibrilledannelse avhenger av at en viss kritisk konsentrasjon av forløperproteinet er oversteget (38). Dette sees for eksempel ved AA-amyloidose hvor det ved tilstander med inflammasjon er økt produksjon av SAA (39) og ved Aß2M-amyloidose hvor det er sviktende utskillelse av ß2M (40).
Forløperen til det amyloide fibrilleproteinet er et autologt protein. Dette er sannsynligvis grunnen til at avleiringene ofte ikke gir opphav til noen lokal immunologisk eller inflammatorisk reaksjon. Avleiringen av amyloid skjer gradvis over år og symptomene kommer som regel sent i forløpet. Organforstørrelse sees f.eks. som hepatomegali og makroglossi, eller amyloide tumores i hud, luftveier og urinveier. Avleiringen kan være massiv, og tilfeller av doblet organvekt er rapportert. Graden av organsvikt er imidlertid ikke alltid korrelert med mengden avleiret amyloid; hvor raskt avleiringene bygges opp og hvor de avleires har større betydning for organfunksjon og prognose enn mengde amyloid i seg selv (41). In vitro studier har vist at enkelte forløperproteiner er vevstoksiske (42, 43).
I all amyloid substans finnes i tillegg til det sykdomsdefinerende fibrilleprotein også en gruppe ekstrafibrillære komponenter, som omfatter serum amyloid P (SAP) (44), proteoglykaner (45, 46), og flere matriksproteiner og glykoproteiner som kollagen IV, apolipoprotein E og laminin. Det er vist i dyremodeller at fravær av disse faktorene vanskeliggjør fibrilledannelse. I tillegg finnes en lite definert faktor kalt «Amyloid enhancing factor» (AEF) som er vist i eksperimentelle modeller å ha betydning for fibrilledannelse (47, 48). Mekanismen for AEFs rolle i fibrilledannelse er ikke klarlagt, men en såkornfunksjon («seeding» på engelsk), som ved prionsykdommer, er foreslått (37).
Flere modeller for fibrilledannelse eksisterer, og en modell som bygger på såkalt nukleeringsteori har stor utbredelse (49, 50). I følge denne modellen er det kritiske steg for avleiring av amyloide fibriller at det dannes en kjerne av amyloide fibriller, som danner et skjelett som nye fibriller bygges på. Videre er det postulert at det kreves en kritisk konsentrasjon av forløperproteinet for å danne prekursorkjerner som akselererer fibrilledannelse (51).
Selv om amyloidavleiring ofte har blitt sett på som en irreversibel prosess, er det mange eksempler i litteraturen på tilbakegang av amyloidose. Ved hjelp av serum amyloid-P-scintigrafi (123I-SAP) har man sett reversibilitet av amyloidavleiringer (52, 53). Tilbakegang av amyloid i flere organer, bedret organfunksjon og økt overlevelse er vist for AA (54) og AL amyloidose (55) etter at mengden av sirkulerende forløpere for fibrilleproteinet er redusert. Mekanismene for nedbryting av amyloid er ikke fullstendig klarlagt. Proteolyse av amyloidavleiringer kan bli indusert av amyloidreaktive antistoffer som vist eksperimentelt i dyremodeller (56, 57). Binding mellom fibrilleprotein og SAP er ved in vitro studier vist å hemme nedbrytning av fibrilleproteinet (58).
3. Diagnostikk
Den diagnostiske prosessen kan summeres i fire trinn:
- Mistanke om amyloidose,
- Verifisering av amyloid,
- Etiologisk diagnose, dvs. typing og
- Avgjøre organutbredelse
En viktig forutsetning for en tidlig diagnose er å ha kjennskap til sykdommen slik at man starter utredning når det er indisert. Når amyloid er påvist er neste skritt å bestemme hvilken type amyloidose pasienten har, og til dette trengs en bred tilnærming. Biopsimaterialet med det amyloide proteinet bør typebestemmes så godt det lar seg gjøre, i første omgang ved immunhistokjemi, helst etterfulgt av massespektrometri. Når type er bestemt må man vurdere om det er en ervervet eller arvelig type. Videre må man utrede pasientens øvrige organer for å finne alle manifeste og subkliniske organaffeksjoner og vurdere om det er systemisk eller lokalisert utbredelse. Organaffeksjon omtales videre i kapittel 4.
3.1 Prøvetaking og påvisning i vev
3.1.1 Prøvetaking
Sikrest og nyttigst informasjon får man ved å direkte biopsere det vevet som er mistenkt affisert. Sikrest fordi sensitiviteten er større, og nyttigst fordi det sikkert definerer at det angjeldende organ er affisert. Hvis dette ikke lar seg gjøre, har indirekte biopsi, ofte fettvevsbiopsi fra abdomen tradisjonelt vært anbefalt (59). Rektumbiopsi har lavere sensitivitet og anbefales ikke lenger som førstevalg ved indirekte biopsi. Svakheten med en indirekte biopsi som fettvevsbiopsi er at den aldri sikkert vil avklare om pasienten har affeksjon av et annet organ man mistenker er affisert, og kun sammen med andre indisier kan sannsynliggjøre dette.
Kirurgisk fettvevsbiopsi (1 cm3) er å foretrekke fremfor fettvevsaspirasjon som har en dårlig sensitivitet i utrente hender (60). En kirurgisk fettvevsbiopsi gir mer materiale, og gjør det lettere å gå videre med ytterligere undersøkelser og typing. Det er viktig å etablere et effektivt samarbeid med kirurgisk poliklinikk (hvis man ikke gjør dette selv), for å unngå at dette forsinker diagnosen. Sensitiviteten for fettvevsbiopsi er vist opptil 80-90 % for AL, men < 15-20 % for ATTR avhengig av sykdomsutbredelse (61-63).
Indirekte biopsi er alltid nødvendig ved autonom nevropati, der det er vanskelig med målrettet biopsi. Ved perifer nevropati er sensitiviteten av fettvevsbiopsi lavere, og biopsi av nervus suralis er da førstevalget ved noen sentra i utlandet. Undersøkelsen er imidlertid for tiden ikke tilgjengelig i Norge.
Nyrebiopsi bør gjøres ved mistenkt nyreaffeksjon, og er ikke forbundet med mer blødningskomplikasjoner enn nyrebiopsi ved andre indikasjoner (64, 65). Benmargsbiopsi er viktig ved mistanke om AL-amyloidose både for å kunne vurdere den underliggende plasmacelleklonen og for å vurdere nedslag av amyloid. Andre organer bør biopseres så fremt svaret gir terapeutisk konsekvens.
Ved hjerteaffeksjon er hjertebiopsi gullstandard. Amyloid fordeles globalt i myokard og sensitivitet og spesifisitet er meget god i trenede sentra. Biopsi kan vurderes fra ikke-affisert organ, og ved hudbiopsi er treffprosent ved ATTRwt ca. 15 %, 45 % ved ATTRv og 70 % ved AL-amyloidose (66). Ved hjelp av amyloidose-skjelettscintigrafi med gradering Perugini 2 og 3 (se avsnitt 3.2.3 og 3.2.5.1) kan man nå i de fleste tilfeller stille diagnosen ATTR-hjerteamyloidose uten biopsi, gitt at AL-amyloidose og MGUS er utelukket. Både MR og ekkokardiografi kan gi mistanke om kardial ATTR-amyloidose, men ikke verifisere amyloid. Den diagnostiske treffsikkerheten ved hjertebiopsi er meget god (67).
Hjertebiopsi utføres ved innstikk i vena jugularis interna, og ved hjelp av Seldingerteknikk legges en hylse ned til overgangen til høyre atrium. Med dette introduseres en biotom som skånsomt føres gjennom trikuspidalklaffen og biopsi tas fortrinnsvis av interventrikulærseptum fra høyre ventrikkel. Det er generelt ca. 1 % risiko for alvorlige komplikasjoner ved diagnostisk hjertebiopsi, imidlertid er den mindre ved hypertrofiske kardiomyopatier som amyloidose hvor perforasjonsfaren er lav.
Man må utvise forsiktighet med organbiopsier ved økt blødningstendens i anamnesen, lave blodplatetall, eller økt INR som kan være tegn på funksjonell faktor X-mangel. I disse tilfellene må det gjøres individuelle vurderinger der man best mulig balanserer gevinst og risiko. De fleste avstår også fra tungebiopsier fordi disse har en tendens til å resultere i langvarig sår- og smerteproblematikk.
Om utredningsbiopsi er negativ eller ved usikkerhet om systemisk eller lokalisert amyloidose, er det nyttig å undersøke om det er tatt andre biopsier fra pasienten og farge disse med kongorødt.
3.1.2 Verifisering av amyloid med kongorødt
Alt amyloid farger positivt med kongorødt, men det kan unntaksvis også annet vev gjøre. For å unngå falskt positivt resultat er det viktig at man også ser grønn, gul eller oransje lysbrytning under polarisert lys (forsidebilde og Figur 2). Uten dette anses ikke fargingen som positiv.
3.1.3 Verifisering av fibriller med elektronmikroskopi
Ved sparsomme avleiringer og ved usikkert resultat av kongorød farging kan elektronmikroskopi brukes for å verifisere amyloid ved påvisning av typisk fibrillestruktur med ikke-forgrenede fibriller på 7-12 nm. Imidlertid er det kun små vevsfragmenter som kan undersøkes med elektronmikroskopi, slik at metoden brukes sjeldent for diagnostikk av amyloid. Elektronmikroskopi er lite egnet til typing av amyloid, men i tvilstilfeller kan immunogull undersøkelse (immunelektronmikroskopi) brukes for å bekrefte f.eks. AL-amyloidose.
3.1.4 Differensialdiagnostikk til andre fibrillære strukturer
En kan finne organiserte deposisjoner i form av fibriller eller mikrotubuli av varierende tykkelse ved elektronmikroskopi i nyrebiopsier ved flere andre sykdommer, som fibrillær glomerulonefritt, immuntaktoid glomerulopati, kryoglobulinemisk glomerulonefritt, og fibronektin glomerulopati. Lysmikroskopisk kan disse ligne på amyloidavleiring, men er vanligvis negative ved kongorød farging. Fibrillær glomerulonefritt viser ofte utvidet mesangium med eosinofilt materiale og ofte fortykkede glomerulære basalmembraner. Immunfluorescens viser vanligvis polytypisk IgG nedslag som bygges opp av ikke-forgrenede fibriller på 10-30 nm ved elektronmikroskopi. De fleste tilfellene er negative ved kongorød farging, men det er rapportert enkelte kongorødt positive kasus. Immunfarging for DNAJB9 har høy spesifisitet og sensitivitet for denne entiteten og sammen med økt fibrilltykkelse ved elektronmikroskopi kan brukes til å utelukke amyloidavleiring.
3.1.5 Immunhistokjemisk typing av amyloid
Amyloidproteinet kan i mange tilfeller påvises i vevsprøven ved immunhistokjemi, hvor flere norske patologilaboratorier kan påvise amyloid A ved AA-amyloidose og immunglobulin lettkjeder ved ALamyloidose. Ved Rikshospitalet gjøres også immunhistokjemi for ATTR-amyloidose. Avdelingen har ikke etablert metoder for immunhistokjemisk typing av andre amyloidoser. Immunhistokjemiske undersøkelser har per i dag god sensitivitet og spesifisitet for AA-amyloidose, mens de for ATTR- og AL-amyloidose har lavere spesifisitet og sensitivitet. Ved større utenlandske sentra, som Mayoklinikken i Minnesota og National Amyloidosis Centre i London, har de senere år andelen systemiske amyloidoser som ikke er av AA- eller AL-type økt betraktelig (fra 1-2 % opp mot 10 %) (3, 68). Denne økningen forklares av at man har innført mer omfattende amyloidtyping utover immunhistokjemi, særlig proteinsekvensering og gentesting. En rekke tilfeller av arvelige amyloidoser har blitt feildiagnostisert som AL-amyloidose (3, 4). En slik feildiagnostisering antas å forekomme også i Norge, og vil kunne utsette pasienten for toksisk behandling uten effekt på grunnsykdommen.
Immunhistokjemisk undersøkelse kan gjøres enten på ufiksert eller formalinfiksert vev, hvor de enkelte patologilaboratorier vil ha sine ulike preferanser. Undersøkelsen gjøres rutinemessig ved lysmikroskopi, men kan også gjøres som immunelektronmikroskopi, oftere med forskningsformål.
3.1.6 Typing ved massespektrometri
Immunhistokjemiske metoder begrenses av hvor sensitive og spesifikke antistoffene er, og at man bare kan evaluere de proteinene man har (gode) antistoffer mot, og det er hovedsakelig AA. Laserdisseksjon med påfølgende massespektrometri (LMD-MS) er en helt annen tilnærming der man dissekerer ut amyloid materiale fra biopsier ved bruk av et lysmikroskop med laserstråle. Bitene som dissekeres fri løses opp og den påfølgende analysen påviser alle proteiner som er til stede i prøven i en viss mengde. Metoden begrenses altså ikke av hvilke proteiner man mistenker eller tester for i utgangspunktet, og gjør at det regelmessig påvises nye proteiner som danner amyloide fibriller. For å verifisere at riktig område med amyloidavleiringer er analysert, må en såkalt amyloidsignatur påvises i tillegg til et dominerende protein som danner amyloidfibrillene. For påvisning av amyloidsignatur kreves deteksjon av minst to proteiner av følgende tre: serum amyloid P, apoAIV og apoE. Massespektrometri regnes nå som gullstandard for typing av amyloid. Metoden er etablert ved Oslo Universitetssykehus i et samarbeid mellom avdeling for immunologi og avdeling for patologi. Avdeling for patologi tar imot vevsprøver fra hele landet for utvidet typing med immunhistokjemi og lasermikrodisseksjon med påfølgende massespektrometrisk typing. Det tas også imot vevsprøver til massespektrometri som viser kongorødt negative avleiringer, ofte med ikke-amyloidogen avleiring av lettkjeder.
LMD-MS kan utføres på alle typer vev, inklusive fettvev (69). Omtrent 40 000-80 000 µm2 mikrodissekert vev trengs for massespektrometrianalyse, f.eks. en vevsmengde svarende til 5-6 glomeruli i et vevssnitt (70). Proteiner i den den kongorødfargede mikrodissekerte vevsbiten brytes ned til peptider. Peptidene fragmenteres så i massespektrometeret og masse og ladning av fragmentioner blir målt (tandem massespektrometri). Disse massedataene brukes til å søke i proteindatabaser for å identifisere proteinene i prøven.
Et massespektrometer med høy masseoppløsning og sensitivitet kan detektere så lite som noen femtomol peptid og normalt identifiseres rundt 50-200 proteiner fra en mikrodissekert prøve. Det amyloide fibrilleproteinet er hovedkomponenten i den amyloide fibrillen og vil i en kongorødfarget vevsbit gi et mer intenst signal i massespekteret enn andre proteiner i prøven. I tillegg til det sykdomsdefinerende fibrilleproteinet finnes også en gruppe såkalte ekstrafibrillære komponenter. Serum amyloid P (SAP) er et av proteinene som via en kalsiumavhengig ligand binder alle typer av fibriller (44). Påvisning av dette proteinet brukes som en markør for mengde amyloid i prøven. Den bør identifiseres som et av de kvantitativt rikeligste proteinene i prøven sammen med fibrilleproteinet. Om SAP-komponenten ikke er til stede, bør man vurdere å ta en ny LMD-prøve for MS-analyse.
Usikkerhet i diagnostiseringen kan oppstå når vevsprøven inneholder blod, da mange potensielt amyloide proteiner finnes normalt i blodet. For å minske andelen bakgrunnsproteiner i prøven er presis mikrodisseksjon av det kongorødfargede området kritisk, og en referanseprøve fra et nærliggende vev med negativ kongorødfarging kan være hensiktsmessig (71).
En stor utfordring for presis typing av amyloidose har vært identifisering av AL-amyloidose. ALpeptidene kan være fra den konstante, den variable eller begge delene av lettkjeden, og peptidene kan være fragmenterte og muterte (72). Proteindatabasen inneholder begrenset informasjon om de variable regioner av immunglobulinene (73), og dette kan gjøre gjenkjenning vanskelig.
En sammenlignende studie av amyloidtyping med LMD-MS og immunhistokjemi viste 100 % konkordans mellom de to metodene i de tilfellene der immunhistokjemi var konklusiv. Imidlertid er immunhistokjemi ikke konklusiv i 20-25 % av amyloidtypinger; og her viste man at LMD-MS økte 17 treffsikkerheten fra 76 til 94 % (74). Mens man ved immunhistokjemi bare kan påvise det man spesifikt tester for, gir LMD-MS mulighet for å påvise typer av amyloidose som det ikke gjøres rutinemessig immunhistokjemi for, som f.eks. AFib, ALECT2, AGel, AApoA1 etc. (75, 76), i tillegg til muligheten for å oppdage proteiner man ikke tidligere visste kunne lage amyloid.
I sjeldne tilfeller kan det påvises flere amyloidproteiner hos samme pasient, enten i samme organ eller forskjellig amyloid i forskjellige organer. Det er oftest ATTR og AL amyloidose som forekommer samtidig (11).
3.2 Utredning med hensyn på organutbredelse ved mistanke om systemisk amyloidose
I tillegg til å typebestemme amyloidet, må man avgjøre om amyloidosen er lokalisert eller systemisk, dvs. hvilke organer som er affisert. Biopsi er gullstandard for påvisning av organaffeksjon. I følgende avsnitt beskrives kliniske og parakliniske undersøkelser som kan styrke mistanke om ulike organaffeksjoner av amyloidose. De enkelte organmanifestasjoner er nærmere beskrevet i kapittel 4.
3.2.1 Klinisk undersøkelse og tegn som kan gi mistanke om amyloidose
Anamnese er viktig. Hvilke symptomer har pasienten og hvor lenge har de vært til stede? Man må spørre detaljert om symptomer på organaffeksjon. Familiehistorie må kartlegges med hensyn på evt. arvelige amyloidoser. Spesielt for AA-amyloidose er det viktig med tidligere sykdommer og rusanamnese som kan disponere for AA-amyloidose (77) (se kapittel 5.2).
I den kliniske undersøkelsen skal man undersøke for affeksjon av de organsystemer som vanligvis rammes av systemisk amyloidose. Husk ortostatisk hypotensjon, karpaltunnelsyndrom, perifer nevropati og tarm-, blære- og ereksjonsfunksjonsforstyrrelser, ødemer og tegn til kronisk inflammasjon og/eller infeksjon. Se etter makroglossi.
Ved mistanke bør man gå videre med målrettet biopsi fra det organ man mistenker er affisert, evt. indirekte biopsi fra annet organ, som fettvevsbiopsi. Amyloidose-skjelettscintigrafi (99mTc-DPD) er anbefalt ved mistanke om ATTR hjerteamyloidose.
3.2.2 Blodprøver og urinprøver
Påvisning av en eventuell monoklonal komponent er sentralt med tanke på AL- og AH-amyloidose. Flere blodprøver og urinprøver er nyttige i utredning av hvilke organer som er affisert av amyloidosen og brukes dessuten i stadieinndeling av AL-amyloidose (se avsnitt 5.1.3).
3.2.2.1 Undersøkelser for monoklonal komponent
Monoklonal gammopati er definert ved funn av monoklonalt immunglobulin (M-komponent) i plasma eller urin produsert av en B-celleklon. Påviste M-komponenter utredes for å gjøre en vurdering av om det foreligger monoklonal gammopati av usikker klinisk signifikans (MGUS), myelomatose, eller lett- eller tungkjedesykdom, herunder AL -og AH-amyloidose (se avsnitt 5.1). Ved påvist MGUS anbefales det vurdering hos hematolog i henhold til utredning skissert i handlingsprogrammet for maligne blodsykdommer (2).
De aller fleste pasienter med AL amyloidose har målbar M-komponent eller patologisk lettkjederatio i serum. Hos alle pasienter hvor AL-amyloidose mistenkes skal følgende undersøkelser gjøres:
- Proteinelektroforese med immunfiksasjon
- Serum frie lette kjeder kappa og lambda (s-FLC)
Dersom ratio kappa/lambda er utenfor referanseområdet, og spesielt hvis >4,0 eller <0,05 tyder dette på en sterkt økt produksjon av den ene lettkjedetypen forenlig med en monoklonal gammopati.
Økning av begge lettkjedetyper, altså med normal kappa/lambda ratio, er typisk for polyklonal immunrespons og sees ofte ved nyresykdom.
Urinelektroforese med immunfiksasjon kan hos enkelte pasienter være nødvendig for å påvise den monoklonale komponenten der man ved måling av frie lette kjeder i blodet ikke har påvist monoklonalitet. Hvis man på annen måte kan følge M-komponenten, er ikke regelmessig urinelektroforese nødvendig. Urinelektroforese med immunfiksasjon tas i morgenurin, men må gjøres som døgnurin hvis det er den eneste kvantitative måten å følge sykdommen på.
3.2.2.2 Andre blodprøver
Andre blodprøver som er aktuelle i utredning og oppfølgning av amyloidose:
- Totalprotein og albumin i serum. Reduserte nivåer kan skyldes tap i urinen, tap i tarm, aktiv inflammasjon eller leversvikt.
- NTproBNP og troponin T (TnT). Hvis en eller begge er over referanseområdet, bør det gjøres ekkokardiografi. Hvis fortsatt mistanke om hjerteamyloidose bør det vurderes å gjøre amyloidose-skjelettcintigrafi. Hjertebiopsi bør gjøres hos unge pasienter, ved Perugini grad 1, MGUS og mistanke om AL amyloidose (se Flytskjema for utredning av hjerteamyloidose i avsnitt 3.2.3).
- Alkalisk fosfatase (ALP) og palpasjon av lever. Hvis en eller begge er forhøyet eller forstørret, og det ikke er annen sikker organaffeksjon, bør leverbiopsi vurderes.
- INR. Hvis uforklart økt bør man vurdere fettvevsbiopsi. Alternative forklaringer til økt INR er leversvikt, warfarinbruk, faktor VII-mangel og vitamin K-mangel.
- SR og CRP kan støtte opp om mistanke om kronisk inflammasjon/infeksjon hvis forhøyet. CRP brukes ofte som surrogatmarkør for SAA i utredning og oppfølging av AA-amyloidose.
3.2.2.3 Urinundersøkelser
Kvantitering av albumin og totalprotein i urinen bør gjøres ved utredning av mistenkt nyreaffeksjon. Albuminuri tyder på glomerulær lekkasje. Totalprotein i urin inkluderer også andre proteiner som filtreres i glomerulus, som immunglobuliner ved monoklonal komponent i blodet (Bence Jones proteinuri), og tubulær proteinuri.
Albumin/kreatinin ratio (AKR) og totalprotein/kreatinin ratio (PKR) bør monitoreres ved oppfølgning av systemisk amyloidose. Selv om døgnurinsamling er den mest presise måten å måle albuminutskillelsen på, gir ikke den økte presisjonen noen klinisk nytte. Vi anbefaler derfor at pasienter følges med spotprøver av urin med kvantitering av albumin/kreatinin ratio. Ved forhøyet ratio bør nyrebiopsi vurderes. Økning av ratioen tyder på økende glomerulær skade og vil således styrke en behandlingsindikasjon.
Mikroskopering av urin er ofte uten anmerkning, mikroskopisk hematuri forekommer; men et nefrittisk sediment er uvanlig.
3.2.3 Hjerteundersøkelser
For å påvise hjerteamyloidose må man kjenne til tegn på sykdommen som bør føre til målrettet utredning. Symptomer ved hjerteamyloidose beskrives i avsnitt 4.1. Ved mistanke om amyloidose med hjerteaffeksjon der påvisning av sykdommen får klinisk betydning for symptomlindrende eller spesifikk behandling, anbefales bruk av flytskjema, figur 3. Flytskjema viser utredning for ATTR og AL-amyloidose med hjerteaffeksjon. Hjerteaffeksjon ved andre typer amyloidose forekommer langt sjeldnere, og påvises ofte ved biopsi av annet organ. Ekstrakardiale såkalte «røde flagg» kan styrke mistanken om om hjerteamyloidose (78).
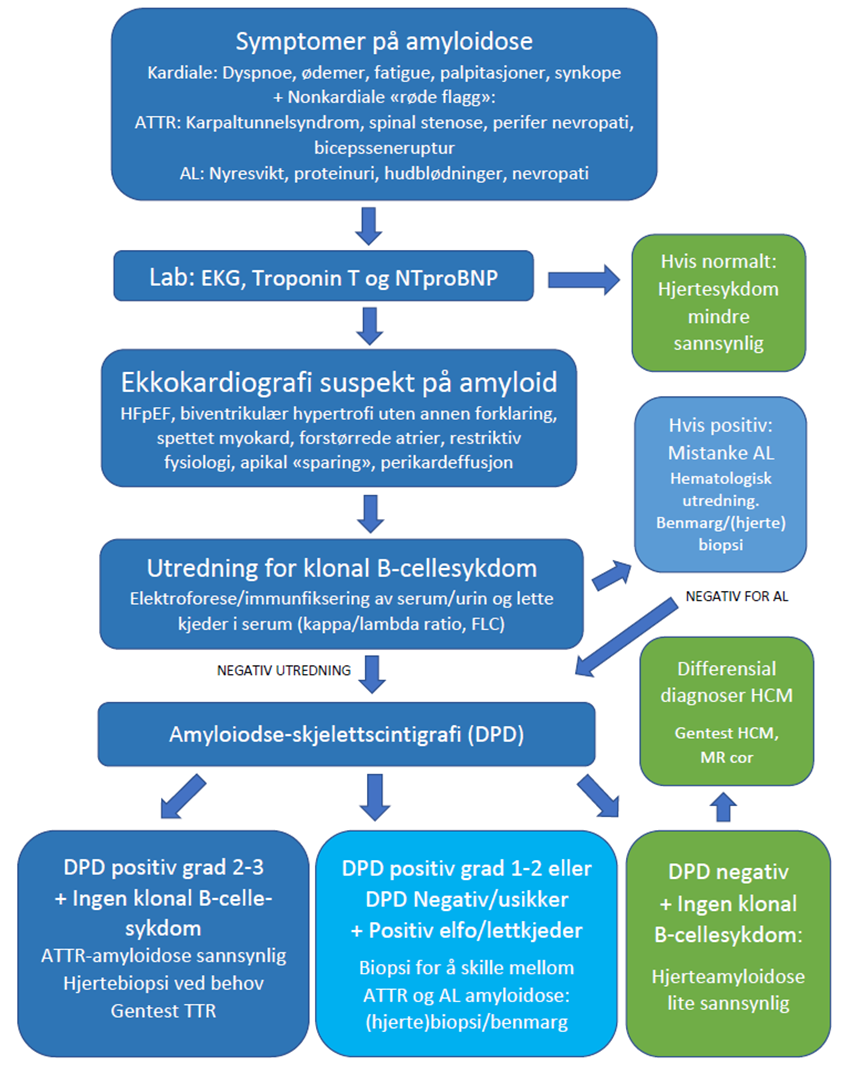
3.2.3.1 Ultralyd av hjertet (ekkokardiografi)
Ultralyd av hjertet er undersøkelsen vi bruker i det daglige pga. god validitet og god tilgjengelighet. Man ser typisk konsentrisk veggfortykkelse (pseudohypertrofi) med affeksjon av begge ventrikler, men kan ikke skille amyloidavleiringer fra andre avleiringssykdommer som Fabrys sykdom, genetisk forårsaket venstre ventrikkel hypertrofi (VVH), eller f.eks. hypertensiv hypertrofi av hjertet. Typisk er septum spettet og fortykket ≥12 mm og oftest også fortykket i bakre vegg. Tykkere septum er forbundet med avansert sykdom og dårlig prognose (79). Venstre ventrikkel har ofte liten kavitet med normal systolisk funksjon og man ser et restriktivt fyllingsmønster med diastolisk dysfunksjon. En ekkokardiografisk spesialundersøkelse (strainmålinger) vil kunne påvise kontraksjons- og relaksasjonsforstyrrelser. Tydelig nedsatt langaksefunksjon i basal- og midtventrikulære deler og nær normal langaksefunksjon apikalt danner grunnlaget for apikal «sparing» som ved hjerteamyloidose har høy sensitivitet jo mer utalt VVH, men er mindre egnet ved lite hjerteaffeksjon. Ved VVH > 15 mm har apikal «sparing» en spesifisitet på 90-95 % og sensitivitet på 80-85 % i trenede hender (80- 82). Det er ofte biatrial forstørrelse og sparsom perikardvæske kan ses hos opptil 50 % av pasienter med AL-amyloidose (83). Ved utvikling av sykdommen vil ejeksjonsfraksjonen etter hvert synke (84).
3.2.3.2 Elektrokardiografi (EKG)
Det er varierende EKG-funn, hvor lav voltage er rapportert i 40-70 % til tross for myokard(pseudo)- hypertrofi med septumtykkelse rapportert > 15 mm. Typiske EKG-forandringer ved hjerteamyloidose er vist i en spansk/italiensk ATTRwt-kohort (85) med lav voltage i prekordial/ekstremitetsavledninger (22 %), lav voltage definert ved Sokolow < 15 mm (49 %), supraventrikulær arytmi (56 %), 1. grads AV-blokk (31 %) og grenblokkmønster, henholdsvis høyre (15 %) og venstre (17 %). Pseudoinfarktmønster var tilstede hos 60 %, mens overraskende kun 11 % tilfredsstilte VVH-kriterier.
15 % av pasienter med AL-amyloidose har atrieflimmer ved diagnosetidspunkt (86). Infiltrasjon av ledningssystemet kan gi AV-blokk grad 2 og 3. 24-timers EKG skal utføres ved svimmelhet, synkope, dårlig pulsrespons eller ved mistenkt arytmi. Ca. 15 % har ventrikulær arytmi, men plutselig død grunnet malign arytmi er sjelden. Redusert hjertefrekvens-variabilitet som tegn på autonom dysfunksjon kan også ses ved 24-timers EKG (87).
3.2.3.3 Høyrekateterisering
Kateterisering av høyre hjertehalvdel gir informasjon om fyllingstrykk og minuttvolum, og informasjon fra denne undersøkelsen kan innvirke på behandlingsstrategi og medikamentvalg (spesielt diuretikabehov). Kateterisering gjøres sammen med hjertebiopsi.
3.2.4 Undersøkelser av mage- og tarmkanalen og lever
Amyloidose med affeksjon av mage- og tarmkanalen gir ofte symptomer som kan ligne andre tilstander. Amyloidose er en aktuell differensialdiagnose i utredning av for eksempel magesmerter og endret avføringsmønster når hyppigere forekommende årsaker, som cøliaki, inflammatorisk tarmsykdom, kroniske infeksjoner og malignitet er utelukket.
Diagnostikk er avhengig av adekvat utredning og prøvetaking. Blødning fra tarmslimhinnen med påvisbart blod i avføring er sjelden i tidlig fase. Amyloidose vil ikke gi utslag på inflammasjonsmarkører som ofte måles i utredning av tarmsykdom, for eksempel fekal kalprotektin (88). Gastrointestinal bildediagnostikk slik som CT abdomen eller MR av tynntarm vil sjelden vise signifikante funn. Ved endoskopiske undersøkelser, kan slimhinnen se helt normalt ut. Det er derfor viktig å ta biopsier til spesialfarging (kongorødt) for å stille diagnosen. Slik farging er ofte ikke rutine ved gastrointestinale biopsier ved norske patologiavdelinger, og samarbeid mellom kliniker og patolog er viktig for å avklare diagnosen. Biopsier fra rektum har ofte vært mest brukt, da de er minst invasive, men amyloidavleiring kan påvises i alle deler av fordøyelseskanalen. Det er viktig med dype biopsier som omfatter submukosa (88).
Funksjonsundersøkelser av øsofagusmotilitet, ventrikkeltømming og tarmmotilitet er ofte patologiske, og kan avklare behovet for tiltak. Ved dysfagi, bør øsofagusmanometri utføres. Ventrikkeltømmingstest er best tilgjengelig med 13C-pusteprøve eller funksjonsultralyd, selv om ventrikkeltømmingscintigrafi fortsatt regnes som gullstandard i utredning av gastroparese (89). En trådløs motilitetskapsel (smart-pill) er også tilgjengelig ved enkelte sykehus. Røntgen transittid kan gi god informasjon om funksjon av tykktarm.
Ved ultralyd av lever kan amyloidose gi et bilde med forstørret lever med heterogen ekkogenisitet, men dette er ikke et spesifikt funn. Ved CT kan det noen ganger ses fokale endringer i attenuasjon eller forkalkninger. MR kan gi økt signalintensitet av leverparenkymet ved T1-vekting (sannsynligvis grunnet småkarsykdom) uten vesentlige endringer ved T2-vekting (90). Diagnosen stilles sikkert kun ved leverbiopsi med spesialfarging. Det er rapportert økt forekomst av blødning etter leverbiopsi ved amyloidose, men dette regnes i praksis ikke for å være et problem ved gode rutiner for observasjon.
3.2.5 Radiologiske og Nukleærmedisinske undersøkelser
Det er to typer aktuelle nukleærmedisinske scintigrafiske metoder i bruk for amyloid-påvisning i Norge i dag: SPECT-baserte ben-avide radionuklider med 99mTc-DPD/HMDP/PYP (se avsnitt 3.2.5.1) som ellers brukes ved skjelettscintigrafi og som kan vise ATTR-hjerteamyloid, og PET-baserte amyloidbindende radionuklider som kan vise beta-amyloid i hjernen (avsnitt 3.2.5.2). MR en aktuell radiologisk billedmetode som kan anvendes i utredning av hjerteamyloidose og ved mistanke om cerebral amyloid angiopati (CAA) (avsnitt 3.2.5.3).
En SPECT-basert amyloid-bindende radionuklide, serum amyloid-P-scintigrafi (123I-SAP), som har sterk affinitet for amyloid (53), er ikke tilgjengelig i Norge i dag. Undersøkelsen ble utviklet for å avbilde systemisk amyloid er bare aktuell hos ytterst selekterte pasienter etter vurdering ved sentra med spesiell interesse for amyloidose. SAP-scintigrafi er tilgjengelig bl.a. ved National Amyloidosis Centre i London.
3.2.5.1 Amyloid-bindende radionuklider ved hjerteamyloidose
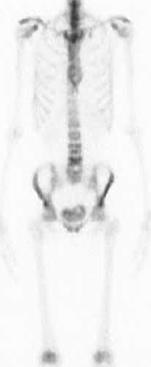
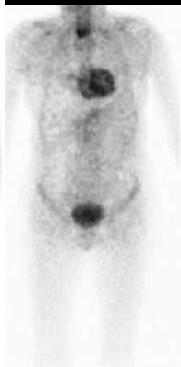
En nukleærmedisinsk helkroppsscintigrafisk ikke-invasiv SPECT-basert billedundersøkelse har i dag en sentral plass i utredningen av ATTR-hjerteamyloidose (91, 92), se flytdiagram for utredning av hjerteamyloidose (Figur 3). I flere tiår har det vært kjent at vanlige skjelettscintigrafi-tracere, bifosfonater (99mTc-DPD (3,3-Difosfono-1,2-propan-dikarboksyl-syre) og 99mTc-HMDP (hydroxymethylenediphosphonate), har høy affinitet for amyloid i hjertet. I tillegg har en mer uspesifikk, men strukturmessig lik tracer (99mTc-PYP, pyrofosfat) høy affinitet for amyloid i hjertet, uten at mekanismen er kjent.
Perugini og medarbeidere viste i 2005 i en mindre studie at 99mTc-DPD scintigrafi har høy sensitivitet og spesifisitet til å påvise ATTR-hjerteamyloidose (83). Fortsatt brukes Peruginis metode for visuell avlesning av 99mTc-DPD- resultatet hvor opptaket i hjertet sammenlignes med opptaket i ben som referanseorgan etter en fire punkts skala (Figur 4). Undersøkelsen omtales i Norge i dag vanligvis som amyloidose-skjelettscintigrafi eller DPD-scintigrafi.
Bruk av skjelettscintigrafi i utredningen av ATTR-hjerteamyloidose ble anerkjent etter en viktig studie i 2016 ved Gillmore og medarbeidere (35). I denne studien, hvor pasientene var henvist på grunn av mistenkt hjerteamyloidose basert på ekkokardiografi og MR, fant man at DPD skjelettscintigrafi har svært høy diagnostisk nøyaktighet. Et positivt DPD-scan med Peruginis grad 2 og 3 har >98% både positiv prediktiv verdi og spesifisitet for å påvise ATTR-hjerteamyloidose, forutsatt klonal B-celle sykdom er utelukket (normal kappa-lambda ratio). Traceren (DPD) tas også opp i hjertet hos en del pasienter med AL-amyloidose, og kan således ikke diskriminere sikkert mellom ulike fibrilleproteiner (93, 94). Det er sporadisk også sett positive funn ved annen arvelig amyloidose som AApoA1 (95). Gillmore viste også at en pasient med negativt DPD-scan (grad 0) uten tegn til lette-kjeder i serum, har svært liten sannsynlighet for hjerteamyloidose. Det må i dag bemerkes at den diagnostiske nøyaktigheten vil kunne endres noe i tiden fremover, hvor målet er å påvise sykdom tidligere og da med mindre sykdomsutbredelse. Det er få pasienter med ATTR-amyloidose i NYHA klasse 1 som er inkludert i studier hvor amyloidose-skjelettscintigrafi har vist meget god treffsikkerhet. Anbefalt indikasjon for amyloidose-skjelettscintigrafi kan være påfallende venstre ventrikkel hypertrofi (>1,2 cm) målt med ekkokardiografi/MR som ikke kan forklares av annen kjent hjertesykdom (hypertensjon, aortastenose, hypertrofisk kardiomyopati (HCM) med mer). Ved bruk av skjelettscintigrafi hos eldre henvist av annen årsak enn amyloidose, f.eks. malignitetsutredning, påvises opptak i hjertet i 1-2 % av pasientene som et bifunn. Det anbefales at alle slike bifunn medfører henvisning til kardiolog med spørsmål om hjerteamyloidose.
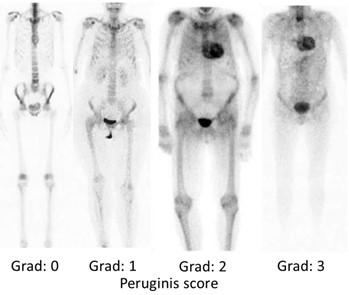
Amyloidose-skjelettscintigrafi gjennomføres ved å injisere 500-700 MBq 99mTc-(DPD/HMDP/PYP). Etter 3 timer gjøres 20 minutter helkroppsavbildning fra isse til underekstremiteter. Ved bruk av DPD og HMDP avleses opptaket etter Peruginis skala, mens ved PYP sammenlignes opptaket mellom hjertet og tilsvarende området på høyre side av thorax, såkalt H/CL (heart/counterlateralt) hvor H/CL>1,5 tilsvarer også høy sannsynlighet for ATTR hjerteamyloidose forutsatt at AL hjerteamyloidose er utelukket ved serumprøve. Dersom Perugini-grad 1-3 eller H/CL ratio>1,5, anbefales et ekstra ca. 15 minutters SPECT/CT opptak over hjertet for å karakterisere både lokalisering og utbredelse av opptaket i venstre og høyre ventrikkel. SPECT vil vise om begge ventrikler er affisert, og om områder i hjertet har lavt opptak. Høyt tracer-opptak i hjertet, sammen med blodprøver som avkrefter AL-amyloidose, har meget høy spesifisitet og sensitivitet for kardial ATTR.
Flere PET-baserte amyloid-bindende radionuklider som brukes til å påvise beta-amyloid i utredning av Alzheimers sykdom (se avsnitt 3.2.5.2) kan også være aktuelle for påvisning av amyloid hjertet, og er godkjent av US Food and Drug Administration (FDA) for klinisk bruk ved utredning av hjerteamyloidose. Imidlertid er foreløpig er PET-basert avbildning for hjerteamyloidose begrenset da undersøkelsen både er mindre tilgjengelig, mindre dokumentert og dyrere enn amyloidoseskjelettscintigrafi (SPECT-baserte).
3.2.5.2 Amyloid-bindende radionuklider ved Alzheimers sykdom
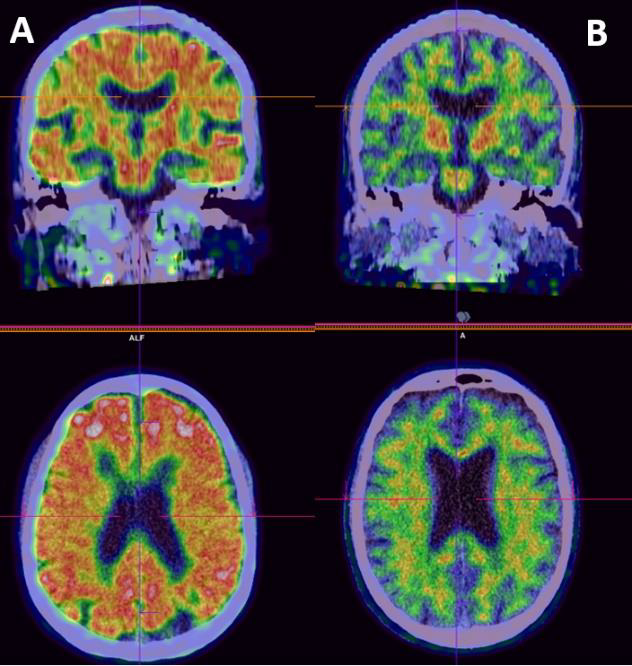
Ved mistanke om Alzheimers sykdom er PET-baserte amyloid-bindende radionuklider som 11C-Pittsburgh compound B (PIB), 18F-florbetapir, 18F-florbetaben og 18F-flutemetamol (Figur 5) i bruk for å detektere amyloide plakk i kortikalt parenkym. I Norge er flutemetamol (18F-Flut-PET) tilgjengelig ved flere sentre.
SPECT-baserte undersøkelser brukes ikke i klinikken for å detektere cerebrale amyloide plakk. Den vanligste metoden for å påvise cerebrale amyloide plakk er spinalpunksjon med påfølgende analyse av amyloid-[beta]-42 (Aβ42). En mer nøyaktig metode er imidlertid å undersøke forholdet mellom 42/40 (96), men Aβ42 analyseres ikke rutinemessig i Norge i dag. Studier har vist at amyloid PET og Aβ42 i cerebrospinalvæske (CSF) er sterkt korrelert (97). 18F-Flutemetamol kan ikke brukes for å påvise amyloid i karveggen, og kan dermed ikke brukes til å diagnostisere CAA. Spinalvæskeundersøkelse er fortsatt førstevalg ved utredning av Alzheimers sykdom. Spinalvæskeundersøkelse er både tilgjengelig, billigere og man vil da, i tillegg til CSF amyloid-[beta], kunne undersøke CSF total-tau og CSF fosfo-tau. Total-tau er uspesifikk markør for nevronskade, mens fosfo-tau er mer spesifikk for Alzheimers sykdom. Patologiske verdier av total-tau og fosfo-tau tilkommer senere i den patologiske prosessen ved Alzheimers sykdom. Hos eldre pasienter med etablert demens og anamnese passende med Alzheimers sykdom, er det ikke nødvendig å bruke spinalvæskeundersøkelse eller radionuklider for å påvise amyloidose.
3.2.5.3 Magnetisk resonans avbildning
MR av hjertet er nyttig i tilfeller der ekkokardiografi viser fortykket vegg og pasienten samtidig har eller har hatt hypertensjon. I disse tilfellene vil MR cor vanligvis gi et sikrere svar enn ekkokardiografi (98). MR gir de mest nøyaktige mål for veggtykkelse og funksjon i venstre ventrikkel og har høyere sensitivitet og kan påvise sannsynlig hjerteamyloidose på et tidligere stadium av sykdommen enn ekkokardiografi (99). Dette har prognostisk verdi. Pasienter med hjerteamyloidose har økt kardialt ekstracellulært volum pga. amyloidavleiring i myokard og kontrastoppsparing langs amyloidnedslag kvantifiserer grad av amyloidmengde. Diffus subendokardial «late gadolinium enhancement» har en spesifisitet på 95 % for hjerteamyloidose (9).
Det finnes protokoller for MR cor både med og uten kontrast, og valg av protokoll vil måtte basere seg på den lokale kompetansen. Det er beskrevet typiske mønstre for «late gadolinium enhancement» (LGE). Ved både ekkokardiografi og MR kan septumtykkelse, atrievolumer og hemodynamiske kriterier brukes som prognostiske markører (100).
Cerebral amyloid angiopati (CAA) er en av de viktigste årsakene til kortikale mikroblødninger og leptomeningeale hemosiderinavleiringer, eller superfisiell siderose. T2*-vektet og susceptibilitetsvektet (SWI) MR brukes for å detektere disse forandringene. SWI er relativt nylig tatt i bruk i rutinediagnostikk og har en betydelig høyere sensitivitet enn T2*-vektede sekvenser. Diagnosen mulig CAA baseres på nevroradiologiske funn ved MR caput og sykehistorie, med støtte i modifiserte Bostonkriterier for CAA-relatert blødning, som beskrevet av Linn og medarbeidere i 2010 (101).
3.2.6 Klinisk nevrofysiologiske undersøkelser
Amyloidose kan både debutere med og etter hvert gi ulike typer perifer nevropati. Dette er særlig hyppig ved AL- og arvelig ATTR-amyloidose (102). Ved mistanke om nevropati enten i form av fokal affeksjon som ved karpaltunnelsyndrom (CTS) eller generell tykkfibernevropati bør pasienter henvises til nevrografi og elektromyografi (EMG) ved et klinisk nevrofysiologisk laboratorium. Hos enkelte pasienter kan både CTS og polynevropatien være asymptomatisk og man bør derfor vurdere å henvise alle pasienter som utredes for spesielt AL- og ATTRv-amyloidose til dette.
Hos pasienter med smerter og/eller autonome symptomer (ortostatisme, urinretensjon, endret svettemønster) hvor man ikke finner patologi ved standard nevrografi og EMG anbefales kartlegging av afferente tynne nervefibre og eventuelt autonome tester. De fleste universitetssykehus gjør Thermotest, en psykofysisk test hvor man tester pasientens evne til å kjenne varme, kulde, varmesmerte og kuldesmerte. I tillegg kan man gjøre hudbiopsi for telling av tynne nervefibre for evt. påvisning av tynnfibernevropati. Avanserte autonome tester er relativt lite tilgjengelig i Norge, men måling av resturin og påvisning av ortostatisme vil kunne gi en pekepinn om autonom dysfunksjon.
Ingen av funnene ved klinisk nevrofysiologi er i seg selv patognomoniske for amyloidose. På den annen side vil bilateral CTS (påvist ved nevrografi) eller en smertefull tynnfibernevropati (Thermotest eller hudbiopsi) som etter hvert også affiserer tykke nervefibre (nevrografi og EMG) støtte opp om diagnosen. Bilateralt CTS regnes som et rødt flagg med hensyn til amyloidose (103) og bør særlig hos menn utløse videre utredning.
3.3 Genetisk utredning ved mistanke om arvelig amyloidose
Muligheten for arvelig amyloidose bør vurderes hos alle pasienter med systemisk amyloidose som ikke har entydig verifisert AA- eller AL-type av amyloidose. Flere av de arvelige formene for amyloidose rammer de samme organer som de vanligst forekommende sporadiske former for amyloidose (Tabell 2). Det er viktig å tenke på arvelige amyloidoser også ved manglende familiehistorie; det kan være nedsatt penetrans eller de novo mutasjoner. Det er lite sannsynlig å finne noe man ikke leter etter, og andelen arvelige amyloidoser er vist å øke ved mer omfattende amyloidtyping utover immunhistokjemi, særlig ved gentesting for kjente mutasjoner og proteinsekvensering (3, 68).
Ved mistanke om arvelig amyloidose bør medisinsk genetiker involveres, se under. Riktig diagnostikk av arvelig amyloidose er viktig, ikke bare for behandling, men også for genetisk veiledning. Ved noen av de arvelige amyloidosene kan det være aktuelt å følge mutasjonsbærere som ennå ikke har utviklet klinisk sykdom med henblikk på å sette inn behandling for å beskytte organfunksjon (f.eks. blodtrykkskontroll, behandling av nyresvikt).
3.4.1 Gentesting
DNA‐analyser er påkrevet for å kunne stille diagnosen arvelig amyloidose. Gentesting for arvelig ATTR-amyloidose gjøres ofte som del av et genpanel ved utredning av kardiomyopatier, og en sjelden gang som rettet analyse (Sanger sekvensering) der det er klinisk mistanke om arvelig ATTR. Disse analysene rekvireres oftest av indremedisiner/kardiolog. Ved genetisk utredning for venstre ventrikkel hypertrofi (VVH) analyseres 77 forskjellige til nå kjente gener, inklusive gener for ATTR.
Andre DNA‐analyser gjøres i svært begrenset grad i Norge i dag. Det vil oftest være naturlig å kontakte medisinsk genetisk avdeling for å få råd og veiledning om testingen. Oppdatert oversikt over hvilke norske laboratorier som tilbyr hvilke tester finnes på www.genetikkportalen.no. Der er også rekvisisjonsskjemaer tilgjengelig. Norske laboratorier videreformidler vanligvis ikke prøver til utenlandske laboratorier; man må sende direkte. I spesialtilfeller kan man be laboratoriet sette opp et individuelt tilpasset(«custom») genpanel som dekker de relevante genene, f.eks. basert på genene som er listet i Genomics England PanelApp (https://panelapp.genomicsengland.co.uk/). Det kan være aktuelt å sende prøvene til spesiallaboratorium i utlandet.
4. Organaffeksjon
Alle kroppens organer kan affiseres av en eller flere typer amyloidose. Ofte vil en pasient kunne presentere seg med symptomer fra og påvist amyloid i ett organ eller organsystem. For å kunne gi riktig behandling er det da viktig å finne ut om det er flere affiserte organer, dvs. systemisk eller en lokalisert amyloidose. Amyloidavleiring kan være uten symptomer fram til alvorlig organsvikt oppstår.
I Tabell 2 listes typiske distribusjonssteder for de vanligste systemiske amyloidosene. Se også Tabell 1. De to organer som er hyppigst rammet ved systemisk amyloidose er nyre og hjerte; dernest lever, mage- og tarmkanalen og perifere nerver. Flere typer amyloidose kan ramme disse organene. Affeksjon av lunger, lymfeknuter, endokrine organer og muskler forekommer også, men er mindre vanlig. Ved avleiring av amyloid i muskler og skjelett mistenkes gjerne AL- og Aβ2M-amyloidose, mens amyloid nevropati taler for AL-, Aβ2M- eller ATTR-amyloidose.
| Hjerte | Nyre | Lever | PNS | ANS | Hud/ bløtvev |
AA | + | +++ | ++ | - | + | + |
AL | +++ | +++ | ++ | + | + | ++ |
ATTRwt | +++ | - | - | - | - | + |
ATTRv | +++ | + | - | +++ | +++ | - |
AFib | - | +++ | + | - | - | - |
AApoA1 | + | + | +++ | - | - | - |
ALECT2 | - | +++ | + | - | - | - |
Aβ2M | - | - | - | - | - | +++ |
AGel | + | + | - | +++ | +++ | +++ |
Tabell 2. Typisk organdistribusjon for utvalgte typer systemisk amyloidose. PNS: perifere nervesystem. ANS: autonome nervesystem. Modifisert etter Palladini og medarbeidere 2020 (104) og Garcia-Pavia og medarbeidere 2021 (78).
De forskjellige kliniske organmanifestasjonene ved amyloidose har likhetstrekk uavhengig av hvilket fibrilleprotein som forårsaket avleiringen. Typiske manifestasjoner for forskjellige organene gjennomgås her i de følgende avsnitt.
Ved systemisk amyloidose er følgende organmanifestasjoner særlig aktuelle:
Hjerte: Restriktiv fylningssvikt (kardiomyopati), hjerterytmeforstyrrelser
Nyre: Nyresvikt, albuminuri evt. med nefrotisk syndrom
Lever: ALP-økning og hepatomegali
Mage/tarm: Blødninger, motilitetsforstyrrelser, avføringsendring, dårlig opptak, vekttap
Nerver: Perifer og autonom nevropati
Lunge: Respirasjonssymptomer
Bløtvev/bindevev: Makroglossi, karpaltunnelsyndrom, blødninger
Skjelettmuskulatur: Svekket kraft, atrofi
Endokrine organer: Svikt i endokrin funksjon
Ved AL-amyloidose brukes ofte følgende kriterier for organaffeksjon ved positiv biopsi fra annet organ (indirekte biopsi) (105):
Nyre: Døgnurin >0.5g protein, hovedsakelig albumin
Hjerte: Gjennomsnittlig tykkelse av septum og bakre vegg > 12mm, uten kjent hypertensjon
Lever: ALP >1,5x øvre referanseområde (eller leverstørrelse >15 cm målt radiologisk i lengdesnitt i midtklavikulærlinjen, uten hjertesykdom) hvis andre årsaker er utelukket/usannsynlige
Lunge: Respirasjonssymptomer + interstitielle forandringer på røntgen eller CT thorax
Bløtvev: Makroglossi
Perifer nevropati: Klinisk, symmetrisk, sensorimotorisk nevropati i underekstremitetene
Autonom nevropati: Motilitetsforstyrrelser i gastrointestinaltraktus, ortostatisk hypotensjon, blæreforstyrrelser
4.1 Hjerte og kar
Ved hjerteaffeksjon avleires amyloidet ekstracellulært og medfører pseudohypertrofi, stivt hjerte med restriktiv fylling, høye fylningstrykk og kan påvirke det elektriske ledningssystemet og klaffene. Hjertet affiseres i ca. 70 % av tilfellene ved AL, hos alle ved villtype ATTRwt og hos 30-100 % ved ulike varianter av arvelig ATTR (ATTRv) (78). Graden av hjerteaffeksjon er den viktigste prognostiske faktoren. Det er som regel biventrikulær global affeksjon. Hjerteaffeksjon sees hos under 10 % ved AA-amyloidose.
Infiltrasjon av amyloid mellom hjertemuskelcellene gir primært en diastolisk dysfunksjon preget av at hjertet blir stivt med redusert relaksasjon og fylning. Ved diastolisk dysfunksjon/relaksasjons-forstyrrelser vil trykket i venstre ventrikkel være høyt, den passive fyllingen skjer da mot lavere gradient og derfor lavere volum. Atrial kontraksjon må derfor stå for en prosentvis større del av fyllingen, og atriene dilateres (106). Ved atriearytmi mistes dette ekstra volumet og pasienten kan få en dramatisk nedgang i minuttvolum. Hjertet kompenserer normalt redusert slagvolum med takykardi og dette medfører en vond spiral som forkorter diastolen og hjertets fyllingsfase ytterligere (107). Amyloide hjerter har imidlertid lavt slagvolum og er avhengig av frekvens for å øke minuttvolum ved anstrengelse, så fylning mot frekvens er sannsynligvis en individuell tilpasning.
Klinisk manifesterer dette seg som tung pust og ødemer deklivt og i tredjerom (perikard, pleura, peritoneum) samt perifere ødemer. Angina pectoris, synkoper og nærsynkoper kan også være presenterende symptomer (67), men koronarsykdom er ikke assosiert med amyloidose. Hvis det ikke samtidig foreligger koronarsykdom, kan angina skyldes økt oksygenbehov fra hjertemuskulaturen. En eldre pasientgruppe kan ha koronarsykdom som uavhengig komorbiditet. Synkoper kan ha sammenheng med ortostatisk hypotensjon og/eller arytmier. Den systoliske funksjonen bevares og reduksjon i denne sees først langt ut i forløpet og er et prognostisk svært alvorlig tegn.
Hypotensjon er et vanlig problem hos pasienter med AL- og ved ATTR-amyloidose, og har ofte flere årsaker. Autonom nevropati, hjertesvikt og medikamentbivirkninger er alle vanlige. I tillegg er rytmeforstyrrelser vanlig, og ved mistanke bør Holterovervåkning utføres. Rytmeforstyrrelser kan forårsake kardiell dekompensering (108). Synkope er et alvorlig tegn, og bradykardi og AV-blokk er en kjent dødsårsak (108).
Ved siden av ATTRwt som deponeres systemisk, men klinisk manifesteres hovedsakelig med symptomer på hjertesvikt, affiseres hjertet ved økende alder av en lokalisert amyloidose dannet fra peptidhormonet atrial natriuretisk faktor (ANF), også kalt atrial natriuretisk peptid (ANP). Fibrillene deponeres kun i hjertets forkamre, som isolert atrielt amyloid. ANF produseres av hjertets forkamre som respons på økt strekk av veggen; og persisterende høy ANF sees ved hjertesvikt (17, 109). Lokalisert AANF sees ved økende alder og hos yngre med hjerteklaffesykdom og kronisk atrieflimmer, og øker sannsynligvis risikoen for å utvikle atrieflimmer (110).
4.2 Nyrer og urinveier
Flere typer amyloidose kan ramme alle deler av nyrer og urinveier. Nyrestørrelsen er initialt normal, og vil senere skrumpe, i motsetning til den organforstørrelsen man ser ved økende amyloidavleiring i en del andre viscerale organer.
Ved glomerulær affeksjon sees albuminuri, som kan være moderat eller manifestere seg som et nefrotisk syndrom kjennetegnet av den klassiske triaden albuminuri, hypoalbuminemi og ødemer. Et viktig poeng ved spørsmål om amyloid nyreaffeksjon er å måle albumin i urinen, i tillegg til totalprotein. Ved AL kan det pga. lette kjeder i urinen (Bence Jones proteinuri) være vanskelig å bruke totalprotein i urin som mål på nyreskade. Albuminuri kan sees hos 97 % av pasientene med AA-amyloidose på diagnosetidspunktet (111) og graden av albuminuri er korrelert med prognose (112). Nefrotisk syndrom med ødemer sees relativt ofte, og nyrevenetrombose og dyp venetrombose er velkjente komplikasjoner som ved nefrotisk syndrom av andre årsaker. Mikroskopisk hematuri forekommer; men et nefrittisk sediment er uvanlig (113).
Gradvis tap av nyrefunksjon er vanlig ved nyreaffeksjon av alle typer amyloidose. Mens AA, AL og AFib-amyloidose ofte presenterer seg med nefrotisk syndrom, er det andre typer amyloidoser, som f.eks. ALECT2, som oftere presenterer seg med gradvis tap av nyrefunksjon uten signifikant proteinuri (114). Mer enn 40 % utvikler endestadie nyresvikt ved AA-amyloidose (111). Nyresvikt og infeksjoner har vært vanlige dødsårsaker ved AA-amyloidose (24, 112).
I urinveiene påvises lokalisert amyloid hyppigst i blæren, etterfulgt av ureter, uretra og nyrebekken. De er oftest av AL-type. Lokalisert amyloid i prostata og sædblære kan sees ved biopsering av disse organene, og er oftest asymptomatisk, men kan gi hematuri eller hematospermi (115). Lokalisert amyloid i blære, ureter og nyrebekken kan klinisk mistolkes som neoplasme og presenterer seg ofte med hematuri (116, 117). Amyloid i urinblæren kan også manifestere seg som livstruende blødning og irritative vannlatingsforstyrrelser.
4.3. Mage- og tarmkanalen
Amyloidose med affeksjon av mage- og tarmkanalen gir ofte uspesifikke gastrointestinale symptomer. Symptomene er forbundet med redusert livskvalitet og dårlig prognose (118). Flere typer amyloidose kan affisere mage- og tarmkanalen, oftest AA, AL eller ATTR (119). Affeksjon av mage-tarmkanalen er rapportert i 3-8 % av pasienter med AL amyloidose (120, 121). Nesten to tredjedeler av pasienter med ATTRv rapporterer symptomer fra fordøyelsessystemet (122), mens ATTRwt sjelden gir slike symptomer.
I tidlig fase av sykdommen er det sjelden vesentlige symptomer fra mage eller tarm, og subklinisk affeksjon kan foreligge. Mekanismene som gir symptomer er ofte sammensatte. Manifestasjoner av sykdommen er relatert til dysfunksjon og skade av det autonome nervesystem, og således kan symptomer i hele fordøyelseskanalen oppstå (118). Det er rapportert både tidlig metthetsfølelse, kvalme, oppkast og vekttap fra øvre deler av fordøyelsessystemet, og vekslende obstipasjon og diare ved tarmaffeksjon. Magesmerter vil ofte være til stede, ofte etter matinntak. Det er imidlertid ingen sikker korrelasjon mellom symptombyrde og alvorlighetsgrad av sykdommen i tidlig fase, og det vil ofte være vekttap og andre almensymptomer som leder oss til diagnosen. I senere stadier av sykdommen forekommer ofte feilernæring og mangeltilstander utløst av malabsorpsjon (123). Massiv gastrointestinal blødning kan forekomme ved uttalt amyloidavleiring som gir skjøre blodkar (124).
4.4 Milt og lever
Ved dyremodeller for AA-amyloidose er milt ved siden av lever vanligvis de første affiserte organene, men gir kliniske symptomer kun ved massiv avleiring sent i forløpet. Ved human AA-amyloidose sees forstørret milt og lever ved diagnosetidspunktet hos 5-10 % (111, 125). Bakterielle infeksjoner forekommer hyppig hos pasienter med uttalt amyloidavleiring i milt (24). Affeksjon av milt med splenomegali er rapportert i 4-13 % av pasientene med AL amyloidose (126).
Det kliniske bildet ved amyloidose i lever er oftest relatert til symptomer fra systemisk amyloidose slik som vekttap og redusert allmentilstand. Leveren er affisert hos 90 % av pasienter med AL amyloidose og 60 % av pasienter med AA amyloidose (127).
Leveren kan være moderat til betydelig forstørret, og ofte er ALP betydelig forhøyet (128). Graden av hepatomegali samsvarer ikke direkte med grad av amyloidavleiring (129). Kronisk leversykdom med leversvikt og portal hypertensjon ved amyloidose i lever er sjelden. Ascites kan forekomme, men er oftere relatert til hjertesvikt og systemisk sykdom enn avansert leversykdom. Leveraffeksjon er imidlertid assosiert med dårligere prognose, og behandlingen retter seg hovedsakelig mot systemsykdommen (128).
4.5 Perifere nervesystem
Nevropati er vanlig ved flere former for ATTR-amyloidose (130). Nevropati rammer også ca. 17-35 % av pasientene med AL-amyloidose, men er her oftest ikke det presenterende symptomet. Den typiske manifestasjonen er en perifer aksonal sensorimotorisk nevropati fulgt av autonom dysfunksjon (hos 65 %) (131). Halvparten har atypiske presentasjoner som f.eks. multifokal nevropati, smertefull nevropati, lumbosakral nevropati (132), overekstremitetsnevropati (133), og kan også ligne på CIDP (Kronisk inflammatorisk demyeliniserende polynevropati) (134). Det er altså vanskelig å ekskludere AL-amyloidose ut fra type nevropati. De generelle symptomene starter typisk med smertefulle parestesier i føttene pga. involvering av tynne nervefibre. Etter hvert affiserer sykdommen også generelle perifere nervefibre, med resulterende nummenhet og muskelsvakhet. De autonome symptomene kan være kvalme, oppkast, tidlig metthet, oppblåsthet, forstoppelse, diaré, ortostatisk hypotensjon, blæreforstyrrelser, svetteforstyrrelser og erektil dysfunksjon.
Alle pasienter med nevropatisymptomer bør vurderes hos nevrolog samt henvises til klinisk nevrofysiologisk undersøkelse for kartlegging av utfall både i det somatiske og autonome nervesystem. Kriteriet for organaffeksjon kan oppfylles ved nevrologiske og klinisk nevrofysiologiske funn og samtidig påvist amyloid annet sted.
4.6 Lunger og luftveier
Både systemisk og lokalisert amyloidose kan affisere lunger og luftveier (135). Diffus interstitiell infiltrasjon med amyloid eller diffus septal amyloidose er vanligvis assosiert med systemisk amyloidose, oftest som manifestasjon av AL amyloidose. Lungeaffeksjon er også hyppig forekommende ved ATTR-amyloidose (136). Siden lungeaffeksjon ved systemisk amyloidose sjelden dominerer det kliniske bildet, diagnostiseres den ofte kun ved obduksjon. For ATTR kan amyloidose-skjelettscintigrafi påvise ekstrakardiell infiltrasjon i f.eks. lunge (137).
Lokalisert amyloid i luftveier er ikke uvanlig og sees i to varianter (138). Trakeobronkial amyloidose debuterer oftest i 50-60-årsalderen. Den kan være asymptomatisk, men viser ofte symptomer på luftveisobstruksjon med stridor, hoste, atelektase og infeksjon. I tillegg finner man regelmessig dyspné, hemoptyse, gjentatte pneumonier og lobær kollaps. Behandlingen er oftest lokal hvor man v.h.a. kirurgi, laser, stråling og stenting prøver å fjerne obstruktive områder. Det er en relativt dårlig prognose med forventet overlevelse mellom 30 og 40 % etter 5 år. Nodulær pulmonal amyloidose affiserer lungeparenkymet som singel eller multiple knuter, oftest påvist som tilfeldig funn. Lokalisert AL-amyloidose i lunger kan være assosiert med Sjøgrens syndrom (139).
Mutasjoner i genet for surfaktant protein C (SPC) er vist å gi lungefibrose med avleiring av amyloide fibriller dannet av SPC (140).
4.7 Hud, bløtvev, bindevev
De fleste former for amyloidose i hud er lokaliserte. Systemisk amyloidose kan imidlertid gi en rekke ulike hudforandringer, som kan være klinisk og histologisk identiske til dem man ser ved lokalisert hudamyloidose. Amyloid i karvegger kan føre til blødninger med purpura, petekkier og ekkymoser, ikke sjeldent periorbitalt. Amyloidnedslag i subkutis som noduli eller plakk kan sees i alle lokalisasjoner, ofte på fleksorsider og i ansikt eller slimhinner. Ved amyloid infiltrasjon av nerver kan man få nevropatiske hudforandringer og sår.
Lokalisert amyloidose i hud uten systemisk affeksjon sees oftest som enten makulær- eller lichenamyloidose, hvor amyloidet er deponert i superfisielle dermis. Fibrilleproteinet er fragmenter av keratin fra degenererte keratinocytter (141). Ved nodulær amyloidose er fibrilleproteinet oftest fra lette immunglobulinkjeder, altså AL, produsert av lokale plasmaceller. Progresjon av nodulær amyloidose til systemisk amyloidose er sjelden, men lokalt residiv er ikke uvanlig (69, 142).
Amyloidomer som består av injiserte proteiner og peptider kan sees ved injeksjonssteder for insulin, hvor det injiserte insulinet danner amyloidfibriller, AIns, og tilsvarende med AEnf-fibriller ved injisert enfurvitide (143).
Bløtvev og bindevev kan affiseres av flere typer systemisk amyloidose. Lumbal spinal stenose, bilateralt karpaltunnelsyndrom (CTS), og bicepsseneruptur er typiske ekstrakardiale manifestasjoner av ATTR (78). Aβ2M som sees ved langvarig endestadie nyresvikt, avleires er først og fremst i bevegelsesapparatet (bløtvev, brusk og ben) med CTS og cystiske benlesjoner (144).
4.8 Øye og øre/nese/hals
Øyemanifestasjoner er relativt sjeldne ved amyloidose, men alle okulære og periokulære strukturer kan bli affisert (145, 146). De hyppigst berørte områdene er konjunktiva og øyelokk, med symptomer som ptose eller oppfylning, men ekstraokulære muskler, tårekjertler eller orbitalt fettvev kan også affiseres. Amyloidose i øyet forekommer både ved lokalisert og systemisk amyloidose, oftest av type AL.
I tillegg kan de første og av og til de eneste symptomene ved sjeldne arvelige former som gelsolin- (AGel) (147), keratoepitelin- (AKer) og laktoferrin-relatert (ALac) amyloidose være oftalmologiske i form av korneadystrofi. Amyloidavleiring i corpus vitreum er kjent manifestasjon ved arvelig ATTRv amyloidose.
Larynx er et av de hyppigst affiserte områdene ved lokalisert amyloidose. Laryngeal amyloidose presenterer seg ofte med vedvarende heshet, men kan manifesteres som dyspnø, hoste eller fremmedlegemefølelse, vanligvis i 50-60 års alder. Forekomsten er lik blant kvinner og menn. Alle områder i larynx kan være affisert: stemmebåndene, samt de supraglottiske- og subglottiske områdene (148). I en studie om 100 pasienter med laryngeal amyloidose (130) hadde en tredjedel avleiringer i flere områder i larynx. Laryngeal amyloidose behandles med kirurgi. I den ovennevnte studien var det nødvendig med flere behandlinger hos ca. halvparten av pasientene pga. residiv.
I sjeldne tilfeller kan laryngeal amyloidose være del av systemisk amyloidose, som AL amyloidose eller arvelig apolipoprotein A1 amyloidose (AApoA1).
4.9 Endokrine organer
Islet amyloid polypeptid (IAPP) eller amylin er et polypeptidhormon som produseres av bukspyttkjertelens β-celler. Ved diabetes type 2 er det vist at IAPP danner amyloide fibriller som avleires lokalt i bukspyttkjertelen; og amyloidavleiringen antas å skade/fortrenge β-cellene og dermed å hemme insulinproduksjonen ved diabetes type 2 (17, 18).
Mikroskopiske amyloidavleiringer i tyreoidea er vanlige ved systemisk AA og AL amyloidose, men massiv amyloidavleiring som forårsaker struma er sjelden. Amyloid struma kan gi luftveisobstruksjon, dysfagi og dysfoni, og kan være assosiert med både hypo- og hypertyreose. Ved medullært tyreoideakarsinom sees ofte amyloidavleiringer bestående av prokalsitonin (ACal).
Amyloidavleiringer i binyre kan ofte påvises ved systemisk amyloidose men gir sjelden binyresvikt, slik at den kliniske betydningen er liten.
4.10 Genitalia og mamma
Amyloidose i vesikulae seminales er en form av lokalisert amyloidose og er sterkt assosiert med aldring. Amyloidproteinet er semenogelin 1, som ofte danner store subepiteliale masser. Amyloidet ved ASem1 diagnostiseres vanligvis som bifunn ved prostatektomi, men kan gi symptomer i form av hematospermi.
Testikulær amyloidose er en sjelden årsak for hypogonadisme og infertilitet. Alle systemiske amyloidoser kan affisere testiklene, men kliniske symptomer er sjeldne. Ved enkelte varianter av ApoA1 amyloidose er testikulær dysfunksjon ofte den første manifestasjonen, spesielt ved Leu75Pro mutasjon. De fleste pasientene med denne mutasjonen er fra et begrenset område i Nord-Italia.
Kvinnelige genitale organer affiseres enda sjeldnere enn mannlige, men det er rapportert lokalisert AL amyloidose i vulva og vagina.
Amyloidavleiringer forekommer sjeldent i bryst. Typisk klinisk presentasjon er en solitær, unilateral, smertefri oppfylning. Amyloidet er oftest av type AL, som kan være del av både systemisk eller lokalisert amyloidose. I en rapport om 40 pasienter med amyloidavleiring i mamma, hadde en tredjedel tegn til ekstranodal marginal-sone lymfom (MALT lymfom) i biopsien i tillegg til amyloid (149).
4.11 Sentralnervesystemet
Flere lokaliserte amyloidoser kan ramme sentralnervesystemet (Tabell 1). Hyppigst forekommende er avleiring av Aβ, som sees ved Alzheimers sykdom (AD) og ved cerebral amyloid angiopati (CAA). Disse tilstandene er nærmere beskrevet i avsnitt 5.7.2. Enkelte systemiske amyloidoser kan også ha avleiring i cerebrale kar eller leptomeninger. Når det gjelder AL, er det her snakk om monoklonal plasmacelleproliferasjon lokalisert i sentralnervesystemet.
Prionsykdommer er sjeldne proteininduserte avleiringssykdommer med dårlig prognose som rammer sentralnervesystemet. Normalt forekommende proteiner mister sin normale konformasjon og avleires i amyloide aggregater. Feilfoldete proteiner kan påvirke normale proteiner og skape en propagering av avleiringene. Mye av patofysiologien er ukjent, men prionsykdommene kan overbringes («smitte») av feilfoldet protein. Eksempler på prionsykdommer i mennesket er Creutzfeldt Jakobs sykdom, Gerstmann-Straussler-Scheinker syndrom, og familiær fatal insomni Prionsykdommene vil ikke bli ytterligere berørt her.
5. De ulike amyloidosene
Amyloidose kan gi opphav til et bredt spekter av symptomer. De ulike fibrilleproteinene (Tabell 1) er assosiert med ulike kliniske uttrykksformer og har typiske predileksjonssteder. Bildet kompliseres imidlertid av at samme fibrilleprotein kan ha ulike kliniske uttrykk og at ulike fibrilleproteiner kan affisere samme organer (150). Identifisering av riktig fibrilleprotein er spesielt viktig slik at man ikke feilaktig gir behandling for en annen amyloidose enn den pasienten har.
5.1 AL-amyloidose
Ved AL-amyloidose produserer klonale B-celler (vanligvis plasmaceller) lette kjeder som felles ut som amyloid ekstracellulært i kroppens vev. Hjerte og nyrer er de organene som oftest er affisert og affeksjon av hjertet er den viktigste prognostiske markøren. Det er viktig at riktig diagnose stilles og behandlingen igangsettes tidlig før irreversibel organaffeksjon oppstår. Behandlingen er per i dag hovedsakelig rettet mot de klonale (plasma-)cellene og ligner på behandlingen av myelomatose (benmargskreft). Hensikten er å forhindre ytterligere produksjon av lette kjeder og dermed forhindre videre organskade. Detaljer rundt behandling av AL amyloidose er beskrevet i nasjonalt handlingsprogram for maligne blodsykdommer (1).
5.1.1 Kliniske manifestasjoner
AL-amyloidose kan gi avleiring av amyloid i alle organer bortsett fra hjerne der kun lokalt amyloidom (151) er beskrevet. Ved påvisning av AL-amyloid i ett organ må man undersøke om det er ledd i systemisk amyloidose (se kapittel 3 og 4 om diagnostikk og organaffeksjoner). Ved systemisk AL-amyloidose er affeksjon av hjerte (60-75 %), nyre (50-70 %), lever (20 %), det autonome nervesystem og mage/tarm hyppigst forekommende (152). Bløtvevsaffeksjon i form av karpaltunnelsyndrom, makroglossi og periorbital purpura er heller ikke uvanlig. Sjeldnere manifestasjoner kan være forkalkninger i milt, fortykket temporalisarterie, struma, prostatatumor, bukspyttkjertelavleiring, spyttkjertelaffeksjon, negledystrofi og bulløse hudforandringer.
Ved affeksjon av hjertet øker NTproBNP tidlig i forløpet. Ekkokardiografi viser typisk restriktiv hjertesvikt med velbevart EF og fortykket interventrikulært septum. Pasientene opplever klassiske hjertesviktsymptomer som tungpust og nedsatt yteevne. Leveraffeksjon gir hepatomegali og stasemønster i blodprøvene med økt ALP og gamma GT, men svært sjelden organsvikt. Nyreaffeksjon manifesterer seg initialt som albuminuri, med påfølgende utvikling til nefrotisk syndrom og gradvis filtrasjonssvikt med synkende GFR. Blødningstendens er ikke uvanlig, og kan skyldes amyloid-avleiringer i små kar og/eller funksjonell koagulasjonsfaktor X-mangel trolig pga. binding av faktor X til amyloid. Blødning rundt øynene skyldes trolig den første mekanismen, og at karene i dette området har lite støtte av subkutant fettvev. Lungeaffeksjon forekommer, men er ikke vanlig ved systemisk AL-amyloidose. Skjelettmuskulatur kan også affiseres. Progressiv muskelsvekkelse bør gi mistanke om dette og det er da indikasjon for muskelbiopsi.
Utredning for AL-amyloidose bør gjøres rutinemessig hos alle pasienter som får påvist klonal B-cellesykdom, og særlig klonal plasmacellesykdom. NTproBNP, albuminuri og ALP bør måles jevnlig for å vurdere om det foreligger begynnende organaffeksjon. Utredning av organaffeksjon summeres i Tabell 3, hentet fra Nasjonalt handlingsprogram for maligne blodsykdommer (1).
Tabell 3. Utredning av organaffeksjon ved verifisert AL-amyloidose: fra Nasjonalt handlingsprogram for maligne blodsykdommer (2)
Hjerte | Alle pasienter Troponin og NTproBNP EKG (lav voltage) Ekkokardiografi (systolisk og diastolisk funksjon, interventrikulær- septum fortykkelse) 24-timers telemetri/Holtermonitorering anbefales til alle med påvist hjerteamyloidose og ved klinisk mistanke om arytmi |
Nyre | Alle pasienter Kreatinin, eGFR, s-albumin U- albumin/kreatinin ratio |
Lever | Alle pasienter Leverfunksjonstester, spesielt ALP Evaluering av leverstørrelse (klinisk eller billeddiagnostisk) |
Tarm | Ved indikasjon Gastro- og/ eller koloskopi med biopsier ved gastrointestinale symptomer inkl. blødning |
Nevropati | Alle pasienter Klinisk evaluering Ortostatisk BT-måling Ved klinisk indikasjon Elektromyografi (EMG) og nevrografi |
Koagulasjon | Alle pasienter INR, APTT. Faktor X måling hvis INR er over referanseområdet eller ved klinisk blødningstendens |
Endokrinopati | Alle pasienter Tyroidea funksjonstester, vitamin D og fastende blodsukker Ved indikasjon Kjønnshormoner |
Lunge | Ved indikasjon Spirometri og røntgen thorax |
5.1.2 Diagnostiske kriterier for AL-amyloidose
For å kunne stille diagnosen systemisk AL amyloidose må alle av følgende fire kriterier være oppfylt (153).
- Histologisk verifiserte amyloidavleiringer med positiv kongorød farging eller amyloide fibriller ved elektronmikroskopi
- Verifisering av AL subtype (samt utelukke andre amyloidformer). Dette gjøres enten ved bruk av immunhistokjemi eller massespektrometri
- Påvist monoklonal plasmacelleproliferasjon ved en eller flere av følgende:
a. M-komponent i serum ved elektroforese/immunfiksering
b. M-komponent urin ved elektroforese/immunfiksering
c. Aberrant kappa/lambda ratio (FLC, dvs. fri lettkjederatio).
d. Klonale plasmaceller i biopsi
- Påvist amyloidoserelatert organskade
Benmargsbiopsi sammen med biopsi av subkutant fettvev påviser amyloid hos 85 % av pasientene. Der disse er negative, og biopsi av affisert organ er betenkelig (f.eks. blødningsrisiko) utføres først spyttkjertel- og/eller rektumbiopsi. Hvis disse biopsiene er negative, og mistanken om amyloidose fortsatt er høy, gjennomføres biopsi av affisert organ.
Immunhistokjemi anvendes primært for verifisering av AL amyloidose, samt for å utelukke andre typer amyloid. Immunhistokjemi er tilgjengelig ved de aller fleste avdelingene for patologi. Imidlertid vil man ved immunhistokjemi i en relativt høy andel av tilfellene ikke kunne klassifisere eller feilklassifisere hvilken type amyloidavleiring som foreligger. Spesifisitet og sensitivitet avhenger av biopsisted og erfaring.
Massespektrometri av biopsimateriale vil i nesten alle tilfeller korrekt klassifisere amyloid-avleiringene. Metoden tilbys som rutineundersøkelse ved Oslo Universitetssykehus (jfr. kap 3.1.6). Biopsier skal sendes til verifisering med massespektrometri ved avdeling for patologi OUS i alle tilfeller hvor konvensjonell histologi/immunhistologi har påvist eller gitt mistanke om amyloidose (uavhengig av type), slik at feilklassifisering og feilbehandling unngås. Unntatt behov for typing med massespektrometri er tilfeller med klar påvist AA-amyloidose, der immunhistokjemien vanligvis er god, og hvor sykehistorien også passer med AA amyoidose.
5.1.3 Stadieinndeling av AL-amyloidose
Prognosen ved AL-amyloidose på diagnosetidspunktet avgjøres særlig av hjerteaffeksjon, nivået av lette kjeder, alder, funksjonsstatus, og antall affiserte organer. Det er utarbeidet flere ulike prognostiske modeller hvor de mest brukte er stadieinndelingen fra Mayoklinikken i Rochester (154), som revideres jevnlig. Revidert Mayo-prognosemodell fra 2012 vises nedenfor (Tabell 4).
Tabell 4. Basert på 810 pasienter sett ved Mayoklinikken innen 90 dager fra diagnose, og validert i to andre materialer på henholdsvis 310 pasienter som gjennomgikk HMAS og 103 studiepasienter fra forskjellige studier (154).
Stadium | Kriterier | Median overlevelse |
I | Ingen av følgende tre kriterier er oppfylt: 1. NTproBNP > 1800 ng/L 2. TnT > 25ng/L (TnI 100 ng/L) 3. dFLC > 180mg/L | NR |
II | Ett positivt kriterium | 68,8 måneder |
III | To positive kriterier | 16,7 måneder |
IV | Tre positive kriterier | 6,7 måneder |
5.1.4 Differensialdiagnoser til AL: Myelomatose og sjeldne plasmacellesykdommer
5.1.4.1 Myelomatose
Myelomatose er den vanligste hematologiske maligniteten med en insidens på ca. 7/100 000 (155). AL-amyloidose er sjeldnere, med insidens ca. 1/100 000 per år (25). Diagnosen myelomatose bygger på biopsi fra benmarg eller biopsi fra en tumor. I tillegg vurderes om det foreligger tegn på organpåvirkning eller tilstedeværelse av myelomdefinerende biomarkører. For detaljer rundt diagnostikk og behandling av myelomatose henviser vi til nasjonalt handlingsprogram for maligne blodsykdommer (2). 10-15 % av pasienter med myelomatose har også AL-amyloidose (156). Når man diagnostiserer en av disse, må man alltid ta stilling til om pasienten også har den andre tilstanden, siden dette har konsekvenser for behandling og prognose.
Hos en pasient med plasmacelleklon og nyresvikt kan det noen ganger være vanskelig å stille den riktige diagnosen. Betydelig albuminuri taler for AL-amyloidose, mens filtrasjonssvikt med kun lett albuminuri taler for myelomatose. Nyrebiopsi vil som regel avklare om den ene eller begge tilstandene er tilstede.
5.1.4.2 POEMS syndrom (Crow-Fukase syndrom, Takatsuki syndrom)
POEMS syndrom er et paraneoplastisk syndrom som skyldes en underliggende klonal plasmacellesykdom. POEMS er et akronym for polynevropati, organomegali, endokrinopati, M-komponent og hudforandringer («skin changes»). Siden også AL-amyloidose kan gi slike funn, er det viktig å kjenne til forskjellene mellom disse sykdommene. I likhet med AL-amyloidose kan man ved POEMS se ødemer, ascites og pleuravæske, mens benlesjoner, Castlemans sykdom, trombocytose og høye nivåer av vaskulær endotelial vekstfaktor (VEGF) er mer spesifikt for POEMS syndrom. POEMS syndrom har nesten alltid lambda-restriksjon, og monoklonalitet med kappa lettkjede vil derfor tale imot POEMS (157).
I likhet med AL-amyloidose, får mange POEMS-pasienter ofte feil diagnose initialt, i én rapport hele 85 % (158). Median tid fra symptomdebut til diagnose er 13-18 måneder (158, 159). POEMS er en meget sjelden sykdom, med en estimert prevalens på 0,3/100 000 (160).
Detaljer om diagnose og behandling av POEMS syndrom gjøres godt rede for i en oversiktsartikkel fra Dispenzieri i 2019 (161). Behandlingen er i hovedsak rettet mot den underliggende plasmacelle-klonen.
5.1.4.3 MIDD – Monoklonal immunglobulinavleiringssykdom
I tillegg til AL-amyloidose finnes det tre andre avleiringssykdommer på bakgrunn av monoklonale B-celler. Til sammen utgjør disse fire MIDD-sykdommene («monoclonal immunoglobulin deposition diseases»). De tre andre er:
- Light chain deposition disease (LCDD)
- Heavy chain deposition disease (HCDD)
- Heavy and light chain deposition disease (HLCDD)
Ved disse sykdommene finner man amorfe kongorødt negative avleiringer, oftest positive for kappa (162) (i motsetning til lambdadominans ved AL-amyloidose). Fragmenter av både lette og tunge kjeder kan være tilstede, og dette definerer de ulike subtypene, den vanligste er LCDD, hvor elektronmikroskopi viser granulære (ikke fibrillære) nedslag. De kliniske syndromene er vanskelige å skille fra AL-amyloidose, da både nefrotisk syndrom, kardiomyopati, leveraffeksjon og nevrologiske symptomer ses. Insidensen er ukjent, men helt klart lavere enn AL-amyloidose. Hvis man utreder en pasient på mistanke om AL-amyloidose med målrettede biopsier, vil immunglobulinavleiringene i disse pasientene oftest kunne påvises.
5.2 AA-amyloidose
AA-amyloidose ble tidligere kalt sekundær amyloidose fordi den kan sees som en komplikasjon til andre sykdommer med langvarig inflammasjon (Tabell 5). Fibrilleproteinet ved AA-amyloidose er amyloid A (163), som er et fragment av serumproteinet serum amyloid A (SAA) (39). SAA er et akuttfaseprotein som produseres hovedsakelig i leveren i respons til proinflammatoriske stimuli, spesielt TNF-alpha, IL-1 og IL-6 (164). Forhøyet serumnivå av SAA over tid ser ut til å være en forutsetning for AA-amyloidose, men er ikke tilstrekkelig i seg selv, da bare en liten andel av pasienter med kronisk forhøyet serumkonsentrasjon av SAA utvikler AA-amyloidose. CRP brukes ofte som en surrogat-markør for SAA, da måling av SAA er lite utbredt i rutinelaboratorier, men CRP har sine begrensninger, f.eks. hos immunsupprimerte hvor SAA ofte øker mer enn CRP (165, 166) og SAA er vist å korrelere bedre enn CRP med sykdomsaktivitet ved revmatoid artritt (167).
Nivået av SAA over tid er korrelert med organskade (168). Median sykdomsvarighet fra debut av grunnsykdom til påvist AA-amyloidose er i en pasientserie fra Nederland vist å være 15 år (spredning 9 mnd-45 år) (125). Prevalensen av AA-amyloidose i ulike samfunn avhenger av prevalensen til de underliggende tilstandene som er ansvarlig for forhøyet SAA, slik som infeksjoner og revmatiske sykdommer.
Tabell 5. Årsaker til AA-amyloidose (utvalgte). Modifisert etter Brunger og medarbeidere (77).
Sykdom/tilstand |
1. Infeksjoner |
Tuberkulose |
Kroniske hudinfeksjoner /injiserende rusmiddelbruk |
Overvekt |
Osteomyelitt |
Bronkiektasier |
Residiverende urinveisinfeksjoner |
Hepatitt B/C |
Cystisk fibrose |
2. Kronisk inflammasjon |
Revmatoid artritt |
Spondyloartropatier |
Urinsyregikt |
Systemisk lupus erytematosus |
Vaskulitter |
Crohns sykdom |
Ulcerøs kolitt |
Sarkoidose |
3. Arvelige sykdommer |
Periodisk febersykdom, inklusiv familiær middelhavsfeber (FMF) |
Immunsvikt (CVID) |
4. Hematologiske sykdommer |
Waldenstrøms makroglobulinemi |
Hodgkins sykdom, Non-Hodgkin lymfom |
5. Svulster |
Maligne: Lungekreft, nyrecellekarsinom |
Benigne: Castlemans sykdom |
6. Idiopatiske |
5.2.1 Årsaker til AA-amyloidose
Ulike årsaker til AA-amyloidose er vist i Tabell 5. En bør utrede pasienten med tanke på å finne den underliggende sykdommen/tilstanden til AA-amyloidose. Diagnosen er av betydning for behandling, oppfølging og prognose. Figur 6 viser forslag til utredning.
Pasienter med inflammatorisk revmatisk sykdom har utgjort en stor andel av pasientene med AA-amyloidose i Norge (169). Inflammatoriske revmatiske sykdommer, spesielt revmatoid artritt, juvenil idiopatisk artritt og spondylartropatier, var bakenforliggende årsak til AA-amyloidose hos 60 % av pasientene i en studie fra London med 374 pasienter med AA-amyloidose (111). I et fransk materiale med 71 pasienter med AA-amyloidose utgjorde pasienter med revmatisk sykdom 31 % (113). Prevalensen av AA-amyloidose hos pasienter med kroniske inflammatoriske revmatiske sykdommer har tidligere vært rapportert opp mot 6 % (170), men ser ut til å reduseres de senere årene (171-173), sannsynligvis pga. bedre immundempende behandling.
Kroniske infeksjoner er hyppigste årsak til AA-amyloidose utenfor Vesten, og er også i Vesten en hyppig årsak. I det britiske materialet med 374 pasienter med AA-amyloidose nevnt over, var kroniske infeksjoner årsak til AA-amyloidose hos 15 % (111), og i det franske materialet utgjorde kroniske infeksjoner 40 % (113). Injiserende rusmiddelbruk med kroniske suppurative infeksjoner er en økende årsak til AA-amyloidose, også i Norge (29). Periodiske febersykdommer er også en viktig årsak til AA-amyloidose (174); i det britiske materialet utgjorde slike sykdommer 9 % av det totale antallet (111), mens i et materiale fra Tyrkia (n=101) utgjorde familiær middelshavsfeber (FMF) hele 81 % av AA-amyloidosene (175). I en studie av nyreamyloidose fra Egypt (n=32) var kroniske infeksjoner årsak til 50 % av tilfellene med AA-amyloidose, hvorav lungetuberkulose var den vanligste årsaken, mens familiær middelhavsfeber var årsak hos 37 % (176). Tilfeller av AA-amyloidose sekundært til familiær middelhavsfeber sees også i Norge, spesielt hos pasienter med bakgrunn fra land rundt Middelhavet.
Andre kjente årsaker til AA-amyloidose er kronisk inflammatorisk tarmsykdom (Crohns sykdom, ulcerøs kolitt), Castlemans sykdom (en polyklonal lymfoproliferativ sykdom der IL-6 er en viktig mediator), visse neoplasmer, f.eks. nyrecellekarsinom, og vaskulitt. I senere år har det vært en økende andel pasienter hvor man ikke finner noen klar underliggende inflammatorisk sykdom (173). Overvekt er nylig vist å være en signifikant risikofaktor for idiopatisk AA amyloidose (177) og AA amyloidose en årsak til nyresvikt hos overvektige (178).
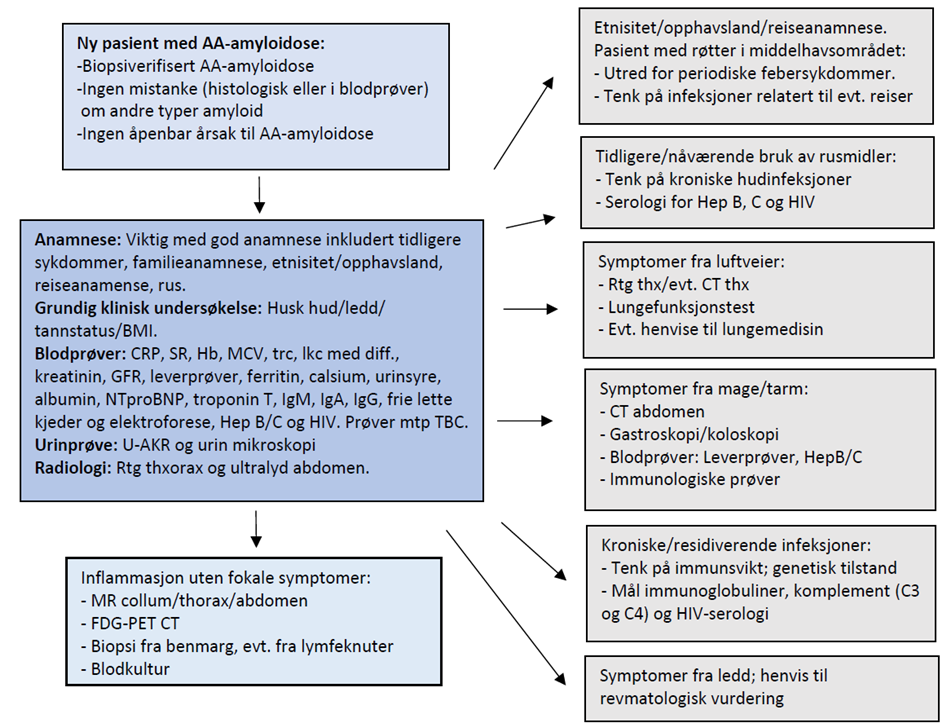
5.2.2 Kliniske manifestasjoner
Amyloidavleiringene vil oftest manifesteres klinisk først når de er ganske uttalte. Symptomer på nyreaffeksjon er det som hyppigst vekker mistanke om amyloidose, og ellers er det generelle symptomer med slapphet og vekttap som dominerer.
Avleiring i nyre, hjerte, mage- og tarmkanalen med lever og milt er vanlig ved AA-amyloidose. Amyloidavleiring i hjerte og lunger forekommer, men er sjelden ved AA-amyloidose (109, 111, 135, 138). Affeksjon av muskler, skjelett eller nevropati er også svært sjelden, makroglossi forekommer ikke. Asymptomatiske AA- amyloidavleiringer er vist i synoviale karvegger. Eksokrine og endokrine kjertler kan være affisert, f.eks. som ved amyloid struma (179).
5.3 ATTR-amyloidose
Transtyretin (TTR) er et amyloiddannende protein både i sin native (normale) form og ved ulike punktmutasjoner, det vil si ATTR-amyloidose forekommer som:
- Ervervet; ATTRwt, hvor wt står for «wild type», på norsk villtype (nativ)
- Arvelig; ATTRv, hvor v står for variant
5.3.1 Ervervet ATTR-amyloidose (ATTRwt)
Normal TTR (TTRwt), kan avleires som amyloid med systemisk distribusjon. Tilstanden ble tidligere ofte kalt senil systemisk amyloidose (SSA), da insidensen øker med alderen (17). Den forekommer oftest, men ikke utelukkende, hos eldre. Vi har i Norge diagnostisert pasienter sent i 50-årene. Nomenklaturkomiteen i International Society of Amyloidosis anbefaler at man ikke lenger bruker begrepet senil systemisk amyloidose, men heller villtype ATTR (12). Symptomgivende ATTRwt sees hyppigst hos menn (180), og er sannsynligvis betydelig underdiagnostisert som årsak til hjertesvikt. ATTRwt har i autopsistudier vært påvist i hjerte og store blodkar hos 20-25 % av personer over 80-års alder og 10 % av personer over 70 år (30, 31).
5.3.1.1 Kliniske manifestasjoner
ATTRwt deponeres hovedsakelig i hjerte, men kan også påvises i mindre mengde i organer som lunger, nyre, lever og milt (181). ATTRwt gir ofte lite og uspesifikke symptomer og kan være vanskelig å diagnostisere (182). Flere tilleggssykdommer hos eldre pasienter bidrar nok også til forsinket diagnostikk av ATTR. I en britisk studie med 102 pasienter med biopsiverifisert ATTRwt var dyspné med diastolisk hjertesvikt vanligste symptom på diagnosetidspunktet (hos 54 %), mens 10 % hadde arytmi som presenterende symptom og 5 % hadde ødemer. Hos 8 % var ATTRwt et tilfeldig funn. Hele 43 % av pasienter med ATTRwt hadde hatt atrieflimmer og 48,5 % hadde hatt karpaltunnel-syndrom oftest 5-10 år før kardial diagnose (182).
5.3.2 Arvelig ATTR-amyloidose (ATTRv)
Arvelig ATTR-amyloidose er knyttet til punktmutasjoner i genet for TTR, og nedarves autosomalt dominant. Over 130 ulike punktmutasjoner i TTR er beskrevet, og de fleste av disse er assosiert med ATTR-amyloidose. Den internasjonale nomenklaturkomiteteen for amyloidose anbefaler bruk av modne proteiner i nummerering av aminosyreresidier, dvs. uten ledersekvens. Nummerering av det fulle forløperproteinet angis vanligvis da i parentes etter det modne proteinet, f.eks. TTRV30M (p.TTRV50M).
Den vanligste mutasjonen ved ATTR-amyloidose er valin erstattet med methionin i posisjon 30 av proteinets 127 aminosyrer, TTR V30M (183). Denne mutasjonen er påvist sporadisk over hele verden, og har endemiske områder i Portugal, Sverige, Brasil og Japan. I Västerbottenområdet i Nord-Sverige er 1 av 50 bærer av denne TTR-mutasjonen, som gir drøyt 7 500 anleggsbærere av sykdommen som også kalles Skelleftesjukan (184-186). Under 10 % av de svenske mutasjonsbærerne utvikler symptomgivende sykdom, og gjennomsnittlig alder ved sykdomsdebut blant de svenske mutasjons-bærerne er 52 år. Pasienter med den samme V30M-mutasjonen i Japan og Portugal har tidligere symptomdebut, gjennomsnittlig 32 år, og nær komplett penetrans (187). Det er ikke kjent hvorfor den samme mutasjonen gir høyere penetrans i Japan og Portugal enn i Nord-Sverige.
Andre eksempler på TTR-mutasjoner som kan nevnes er V122I som sees hos ca. 4 % av afro-amerikanere (188), og er assosiert med økt risiko for hjertesvikt (189). Mutasjonen L111M, også kalt dansk mutasjon, gir restriktiv kardiomyopati rundt 40-års alder (190). TTR-mutasjonen L55P har en svært rask og aggressiv sykdomsutvikling med omfattende amyloidavleiring og symptomdebut med polynevropati rundt 20-års-alder (191). Prevalensen av ATTRv i Norge er ikke kjent, men vi kjenner til enkelttilfeller med arvelig ATTR-amyloidose i flere regioner i Norge. Alle disse pasientene er av utenlandsk opprinnelse og noen egen «norsk» founder-mutasjon er hittil ikke påvist. Det kan være vanskelig å skille ATTRwt fra ATTRv når ATTRv debuter i høy alder. Det er ikke alltid positiv familiehistorie, og det kan være aktuelt å genteste enkeltkasus med ATTR-amyloidose for relevante mutasjoner.
5.3.2.1 Kliniske manifestasjoner
Arvelig ATTR-amyloidose kalles ofte familiær amyloid polynevropati (FAP) fordi de vanligst forekommende mutasjonene særlig affiserer perifere nerver og typisk manifesteres ved progressiv, alvorlig sensorisk/motorisk nevropati. Dessuten affiseres autonome nerver med mage/tarm- og blære-dysfunksjon, impotens og ortostatisk hypotensjon. Gastrointestinale symptomer starter ofte med obstipasjon og oppkast, senere diaré og underernæring. Hjerteaffeksjon er mindre vanlig ved V30M-mutasjonen, men vanlig ved enkelte andre mutasjoner, og der hjerteaffeksjon er presenterende og dominerende symptom, brukes ofte betegnelsen familiær amyloid kardiomyopati (FAC). Typisk sees en restriktiv kardiomyopati som utvikles over mange år. I tillegg til FAP og FAC er det påvist ATTR-varianter assosiert med amyloid nefropati og opasiteter i corpus vitreum, lepto-meninger og nyrer (187, 192). Flere ulike kombinasjoner av organmanifestasjoner ved ATTR-amyloidose er beskrevet i familier over hele verden.
5.4 Andre arvelige amyloidoser
Arvelige amyloidoser skyldes mutasjoner der sykdommen oftest nedarves autosomalt dominant. Amyloidosen skyldes livslang produksjon av et protein som har tendens til å danne amyloid. For oversikt over påviste mutasjoner ved arvelig amyloidose vises til databasen «Mutations in hereditary amyloidosis» som drives av det britiske nasjonale amyloidosesenteret i London (193). Arvelige systemiske amyloidoser er en heterogen sykdomsgruppe med ulikt deponeringsmønster og penetrans. Det kliniske bildet, debutalder og prognose varierer sterkt blant bærerne av samme mutasjon, noe som tyder på at andre faktorer (genetiske, miljøfaktorer) kan ha etiologisk og patogenetisk betydning.
Den vanligst forekommende arvelige amyloidose er ATTR, som er beskrevet i forrige kapittel. Andre proteiner assosiert med arvelige amyloidoser er listet i Tabell 1, bl.a. fibrinogen, apolipoprotein AI og AII, lysozym, gelsolin og cystatin C. Det er også påvist arvelig variant, dog svært sjelden, av både AL- (194) og β2M-amyloidose (195). Selv om AA-amyloidose i noen tilfeller kan være følge av en arvelig sykdom, som familiær middelhavsfeber eller andre autoinflammatoriske syndromer, er AA-amyloidose i seg selv ikke arvelig. De arvelige amyloidosene er sjeldne, men noen former er relativt vanlige i visse geografiske områder.
5.4.1 Fibrinogenamyloidose (AFib)
Fibrinogen er et plasma glykoprotein som syntetiseres i lever. Det har tre strukturelle subenheter, hvor amyloiddannelse er assosiert med mutasjoner i fibrinogen alfakjede (196). Fibrinogen-amyloidose rammer spesielt nyrer, men er beskrevet også i lever og perifere nerver. AFib er den vanligste arvelige årsaken til nyreamyloidose i Storbritannia (196). Deponeringsmønsteret ved AFib er karakteristisk med lokalisering nesten utelukkende i glomeruli og minimal affeksjon av kar og interstitium (114). I en gjennomgang av 71 pasienter med AFib ved National Amyloidosis Centre i London hadde de fleste pasientene ingen familiehistorie på amyloidose, og det ble funnet svært beskjeden amyloidavleiring i andre organer enn nyrer (196). AFib kan gi raskt progredierende nyresvikt og prognosen med nyreerstattende behandling er sammenlignbar med aldersjusterte individer med ikke-diabetisk nyresvikt. De siste årene er fibrinogenamyloidose påvist hos et fåtall norske pasienter (197).
5.4.2 Apolipoprotein A og apolipoprotein C amyloidose (AApoAI, AApoAII, AApoAIV, AApoCII og AApoCIII)
I likhet med serum amyloid A, som er et apolipoprotein, er både apoAI, apoAII, apoAIV, apoCII og apoCIII vist å kunne danne amyloide fibriller som avleires med systemisk deponeringsmønster. Amyloidose fra apoAIV regnes per i dag som en ervervet amyloidose (198) og er påvist i Norge. De øvrige (apoAI-, apoAII-, apoCII- og apoCIII- amyloidose) er arvelige, og assosiert med en rekke ulike mutasjoner som nedarves autosomalt dominant (199-202). Vi kjenner ikke til norske tilfeller av amyloidose knyttet til disse variantene.
ApoAI og apoAII syntetiseres i lever og tynntarm, mens apoAIV syntetiseres hovedsakelig i tynntarm. ApoAI er vist å gi amyloidose i lever, nyre, larynx, hud og myokard (199), distribusjonen er således svært lik den ved AL-amyloidose (Tabell 2). Levertransplantasjon kan hindre progresjon av amyloidnedslagene, og bør iverksettes før sykdommen har medført irreversibel organskade (199). For AApoAII-amyloidose er nyresvikt det dominerende kliniske uttrykk (200). Nyretransplantasjon er ofte vellykket, og det er ingen spesifikk behandling. ApoAIV er hovedsakelig produsert av tynntarmen, og AApoAIV-amyloidose er beskrevet hovedsakelig i nyremargen med langsom utvikling av nyresvikt (198). ApoAIV er også del av den såkalte amyloidsignaturen og finnes i nesten alle amyloidavleiringer, men i mye lavere intensitet og andel enn ved AApoAIV amyloidose. Mutasjoner i apolipoprotein C II og CIII er også vist å forårsake systemisk amyloidose med alvorlig nyreaffeksjon (201, 202).
5.4.3 Gelsolin-amyloidose (AGel)
Gelsolin-amyloidose, AGel, ofte kalt familiær amyloidose av finsk type (FAF), fordi den først ble beskrevet og er hyppigst sett i Finland (203), men samme mutasjon (gelsolin Asp187Asn) og kliniske bilde er beskrevet i flere andre land (114). Gelsolin inngår i aktinmetabolismen. Ved denne mutasjonen sees de første symptomene vanligvis i 25-35 års alder med affeksjon av hornhinnen (hornhinne-gitterdystrofi), gradvis utvikling av polynevropati som rammer hjernenerver og perifere nerfer, typisk med med bilateral facialisparese nedsatt sensibilitet i ansikt grunnet affeksjon av n. trigeminus, dysartri og tungeatrofi ved affeksjon av n. glossofaryngeus og n. hypoglossus, samt perifer polynevropati og autonom dysfunksjon med bl.a. ortostatisk hypotensjon (147). Hudforandringer er vanlig, spesielt cutis laxa (147). Hjerteaffeksjon forekommer, og i senere stadier ses ofte nyreaffeksjon med proteinuri (147, 204, 205). Flere mutasjoner i gelsolin er påvist, både en dansk type (gelsolin Asp187Tyr) hvor fenotypen er svært lik den finske typen, og to øvrige mutasjoner med ren nyreaffeksjon (206, 207). Det finnes ingen spesifikk behandling, men mange må ha gjentatte kirurgiske inngrep for plager fra øyne, nerver og hud. AGel gir vanligvis ikke forkortet livslengde (147).
5.5 ALECT2-amyloidose
Fibrillene dannes fra serumproteinet leukocytt kjemotaktisk faktor 2 (LECT2), som produseres i lever (208-211). Årsaken til amyloiddannelsen er ukjent, og ingen dokumentert behandling foreligger (210). Det er ikke påvist mutasjoner i LECT2 genet, selv om sykdommen kan være assosiert til en singelnukleotid-polymorfisme (SNP) i posisjon 172 (210). LECT2-amyloidose er første gang beskrevet i 2008 (212). I et amerikansk materiale med 414 nyrebiopsier med amyloidose (210), fant man 40 tilfeller av ALECT2. Bare nyreaffeksjon av AL-amyloidose hadde hyppigere forekomst. Tilstanden rammer oftest pasienter av spansk herkomst og påvises i denne befolkningsgruppen i opptil 50 % av pasienter med påvist nyreamyloidose. I vår erfaring er ALECT2 amyloidose sjeldnere i Norge enn internasjonalt, med under 1 % av prøvene i de siste årene. Årsaken til lavere forekomst er ukjent, men etnisitet og sammensetning av populasjonen kan bidra. Det er usikkert om det er familiær opphopning. I det amerikanske materialet var det ett brødrepar med ALECT2 (210).
ALECT2-amyloidose rammer klinisk særlig nyrer, men deponering er også påvist i lever, hjerte, benmarg, prostata, og tarm (211, 213). ALECT2-amyloid i nyrebarken dominerer, med involvering av nyremarg i bare ca. 1/3 tilfeller. Klinisk presentasjon er vanligvis kronisk nyresvikt. Nefrotisk syndrom er sjelden (114).
5.6 Aβ2M-amyloidose
β2-mikroglobulin er et HLA klasse I-molekyl som er tilstede på overflaten av alle kjerneholdige celler. β2-mikroglobulin-amyloidose, Aβ2M, er en komplikasjon til endestadie nyresvikt som resultat av defekt utskillelse av dette proteinet. Aβ2M sees hos pasienter med alvorlig nyresvikt, oftest etter flere år i dialysebehandling, og kalles derfor ofte dialyse-relatert amyloidose. Sykdommen antas å skyldes at dialysen ikke er i stand til å fjerne β2-mikroglobulin fra plasma og at nyresvikten medfører redusert renal utskillelse av proteinet (40, 214).
Forekomsten av Aβ2M er korrelert med hvor mange år pasienten har vært i dialyse og er i studier fra 1990-tallet påvist hos 1/3 som har vært i dialyse i 2-4 år, og 9/10 som har vært i dialyse 7-13 år (215). Nyretransplantasjon vil normalisere serum-nivåene av β2-mikroglobulin, og dermed bremse sykdomsutviklingen. Forekomsten er fallende i senere år med mer effektiv dialyse (216).
Amyloidet er systemisk utbredt, men predileksjonssteder for Aβ2M er først og fremst i bevegelses-apparatet (bløtvev, brusk og ben). Den første manifestasjonen er vanligvis karpaltunnelsyndrom, samt diffuse artralgier, særlig i skuldre. Dessuten sees artropati og cystiske benlesjoner; av og til patologiske frakturer. Spondyloartropati kan også forekomme, særlig i cervikalcolumna (144).
Det er også påvist en sjelden form for arvelig β2-mikroglobulinamyloidose, Aβ2M D76N, som ikke er assosiert med nyresvikt, men hvor mutasjonen i β2M gjør proteinet svært tilbøyelig til å danne amyloide fibriller, og hvor først og fremst autonome nerver rammes (195, 217).
5.7 Lokaliserte amyloidoser
Amyloidavleiring som er begrenset til kun ett organ eller organsystem, kalles lokalisert amyloidose. Lokalisert amyloid kan bestå av multiple avleiringer i samme organsystem. Lokalisert amyloidose er ofte assosiert med aldring. Eksempler på lokalisert amyloid som er vanlig forekommende er AMed i aortaveggen som er til stede hos de fleste etter 50-årsalder (218), Aβ-amyloid i hjernen ved Alzheimers sykdom (15), og AIAPP i de Langerhanske øyer ved type 2 diabetes (18). Fibrilleproteinet ved lokalisert amyloid ser ut til ikke å ha en sirkulerende forløper, men er dannet lokalt. Nodulær lokalisert amyloidose kan gi tumormistanke, f.eks. i lunge, larynx, og urinveier, og blir ofte biopsert.
Noen av de systemiske amyloidosene kan også opptre i lokaliserte former, med lokalt produsert fibrilleprotein, for eksempel lette kjeder fra plasmaceller. De lokaliserte amyloidosene har som oftest en langt mindre alvorlig prognose enn sin systemiske motpart. Andre lokaliserte amyloidoser har ikke en systemisk motpart, og vi skiller gjerne mellom disse to gruppene, altså
- Lokalisert variant av fibrilleprotein som også kan gi systemisk amyloidose og
- Ren lokalisert variant
Ofte kan lokaliseringen til amyloidet gi en pekepinn på hva slags amyloidose det er snakk om. Lokaliserte amyloidoser, oftest av AL-type, viser histologisk ofte rikelig med kjempeceller av fremmedlegemetype ved amyloidet (219). Lokaliserte varianter av tungkjedeamyloidose, AH, forekommer på samme måte som for AL, men er svært sjeldne. Lokalisert variant av AA og Aβ2M er beskrevet, men forekommer også svært sjelden.
5.7.1 Lokalisert variant av AL
AL-amyloidose er den vanligste lokaliserte amyloidosen. Lokalisert AL-amyloidose er ikke et resultat av en underliggende systemisk plasmacellesykdom, men kan være en respons til lokal inflammatorisk reaksjon med klonal plasmacellereaksjon, f.eks. i urinveiene (220).
Larynx, lunge, urinblære, og hud affiseres hyppigst av lokalisert AL amyloidose. Hvis det er lokal AL amyloidose uten tegn til systemisk affeksjon, vil disse svært sjelden utvikle seg til systemisk AL amyloidose. De må imidlertid følges med tanke på evt. lokalt residiv som ikke sjelden forekommer.
Lokalisert AL kan ses i hud som asymptomatiske plakk, fissurer eller noduli. Lokalisert AL kan også sees i urinveier, luftveier, bryst, lymfeknuter, tarm, orbita og i hjerne (121, 149, 221-225). Ofte forekommer lokalisert AL som en tumor, evt. som multiple noduli, som i noen tilfeller er kalsifiserte. Hos disse pasientene vil man ikke kunne påvise en monoklonal komponent i serum eller urin, i så fall må man mistenke systemisk amyloidose (226). Lokalisert AL har som oftest en god prognose, utvikler seg nesten aldri til systemisk sykdom og behandles ikke med kjemoterapi (226).
5.7.2 Lokalisert amyloid i sentralnervesystemet
I sentralnervesystemet avleires amyloid-[beta] (Aβ) som amyloide plakk i det kortikale parenkymet samt i veggen på arterier, arterioler, vener, og kapillærer. Alzheimers sykdom kjennetegnes av gradvis økende kognitiv svikt. Ved Alzheimers sykdom avleires Aβ42 i hovedsak i parenkymet, mens ved cerebral amyloid angiopati avleires hovedsakelig Aβ40 i karveggen. I praksis ser man at det er betydelig overlapp i avleiringsmønsteret ved de to tilstandene. Cerebral amyloid angiopati kjennetegnes av mikroblødninger i det kortikale parenkymet, eller superfisiell hemosiderose, dvs. rester av blodprodukter subaraknoidalt. Både mikroblødninger og superfisiell hemosiderose identifiseres best ved spesielle MR-sekvenser som er følsomme for hemosiderin. CAA kan også føre til parenkymblødninger. Diagnosen CAA stilles ved hjelp av de modifiserte Bostonkriteriene (101). Diagnosen krever samsvar mellom klinikk og typiske funn på MR.
Prevalensen av amyloid i hjernen hos personer med normal kognitiv funksjon varierer fra 3-10 % ved 50-60 års alder, til 40-44 % ved 80-90 års alder (227). Personer med påvist amyloid i hjernen har 2,6 x økt risiko for å utvikle AD sammenlignet med personer uten (228).
Diagnosen Alzheimers sykdom er ikke forenlig med normale verdier av CSF Aβ42 og/eller normal cerebral amyloid-PET. Dog kan amyloide plakk forekomme opptil flere tiår før identifiserbar kognitiv svikt, og biomarkørevidens for cerebral amyloidose må ikke alene brukes til å stille diagnosen. Undersøkelse med amyloid-PET er ikke egnet eller indisert til å identifisere amyloid i kar, og selv om patologiske konsentrasjoner av CSF Aβ40 settes i sammenheng med CAA (229), er ikke dette en etablert metode i klinikken.
6. Behandling av amyloidose
Mange pasienter har sammensatte symptombilder, med affeksjon av flere organsystemer. Et godt tverrfaglig samarbeid i utredning, behandling og oppfølging er viktig. Ulike systemiske amyloidoser krever ulik behandling. Derfor er en mest mulig presis diagnostikk nødvendig. For noen av amyloidosene finnes det spesifikk behandling, for andre ikke.
6.1 Spesifikk behandling rettet mot det amyloiddannende protein
Behandling som virker ved målrettet å redusere tilgjengelig mengde av det proteinet som danner fibrillene, er godt etablert og er spesifikk for hver enkelt type amyloidose. For eksempel vil AL-amyloidose, som skyldes en klonal plasmacellesykdom, behandles med kjemoterapi og evt. høydose kjemoterapi med autolog stamcellestøtte (HMAS), mens AA-amyloidose behandles ved å redusere inflammatorisk aktivitet, da SAA er et akuttfaseprotein. Arvelige amyloidoser kan behandles med levertransplantasjon i de tilfeller der leveren er ansvarlig for syntese av variant form av den amyloiddannende protein (f.eks. ATTRv). I senere år er det også utviklet medikamenter som kan hemme syntese av fibrilleproteinet på gennivå, se mer under avsnittet om TTR-syntese-hemmende behandling.
Et annet angrepspunkt for å redusere tilgjengelig fibrilledannende protein er å stabilisere proteinet i en struktur der det ikke danner fibriller. Slik terapi er etablert for ATTR, se avsnitt 6.1.3.2 om konformasjonsstabiliserende behandling.
6.1.1 Systemisk AL-amyloidose
Prognosen ved systemisk AL amyloidose avhenger av organaffeksjon og størrelsen på B-celleklonen. Amyloid hjerteaffeksjon er den viktigste prognostiske faktoren (230, 231). Pasienter med NYHA stadium III hjertesvikt har en median overlevelse på fire måneder (232), mens median overlevelse hos hjertefriske pasienter med perifer nevropati er mer enn fem år (152). Prognosen ved AL-amyloidose er bedret de senere år grunnet nye og bedre behandlingsmuligheter.
Behandling for systemisk AL-amyloidose handler om å redusere B-celleklonen (oftest en plasma-celleklon) ved bruk av kjemo- og immunterapiregimer, for å minske og helst stanse produksjonen av det amyloidogene proteinet. Hvis dette lykkes, kan man etter måneder og år se økende organrespons i form av funksjonsbedring og bedring av målbare parametere (152). Toleransen for behandling er ofte dårligere ved amyloidose enn ved myelomatose pga. organaffeksjon og medikamentene må justeres avhengig av dette. Når det gjelder detaljer rundt behandlingen vises det til Nasjonalt handlingsprogram for maligne blodsykdommer (1, 2).
6.1.1.1 Førstelinjebehandling
Behandling bør startes straks diagnosen er stilt med sikkerhet, prognosegruppen er avklart og komorbiditet er klarlagt. HMAS er anbefalt 1. linjebehandling til pasienter som vil tåle dette med god mulighet for langtidsoverlevelse. I en nylig publisert randomisert studie hos pasienter med nyoppdaget AL-amyloidose har tillegg av det anti-CD38 monoklonale antistoffet daratumumab til konvensjonell behandling vist dypere og bedre respons (233). For pasienter som er mulige HMAS-kandidater anbefaler vi per desember 2021 induksjonsbehandling med 4 sykluser Daratumumab + Cyklofosfamid, Bortezomib og Dexametason (Dara-CyBorDex).
For pasienter som ikke er høydosekandidater anbefales behandling med Dara-CyBorDex, alternativt kan MelBorDex eller CyBorDex brukes hvis daratumumab ikke er tilgjengelig. Kombinasjonen daratumumab-CyBorDex er til vurdering i Nye Metoder.
Behandlingsmål er minst «very good partial response» (VGPR) tre måneder etter HMAS eller etter fire sykluser med kjemoimmunterapi (234). Pasienter som ikke oppnår dette bør vurderes for å endre behandling. Det samme gjelder hvis det ikke oppnås partiell respons (PR) etter to sykluser. Plasmacellebyrden er som regel lav ved AL amyloidose slik at det ikke er nødvendig med like lang behandling som ved myelomatose. Pasienter med <10 % plasmaceller i beinmargen vil ofte klare seg med 4-6 behandlingssykluser.
6.1.1.2 Høydosebehandling med autolog stamcellestøtte (HMAS)
For pasienter uten alvorlig organdysfunksjon er HMAS fortsatt aktuelt. Vi mangler imidlertid randomiserte studier som støtter denne anbefalingen. Det er bare gjort en fase-III-studie som sammenlignet høydose cytostatika-behandling med autolog stamcellestøtte (HMAS) med Melfalan-Dexamethason (Mel-Dex) hos pasienter under 65 år. Hovedfunnet var at det ikke var noen forskjell i overlevelse mellom gruppene (235). Imidlertid var transplantasjonsrelatert mortalitet i studien svært høy (24 %), noe som er langt høyere enn dagens nivå på ca. 5 %. Sannsynligvis var seleksjonen av pasienter til studien ikke optimal. Flere retrospektive analyser har vist at HMAS hos nøye utvalgte pasienter gir god og langvarig respons og overlevelse (236-239).
Induksjonsbehandling med fire (opptil seks) sykluser Daratumumab-CyBorDex før høydose-behandlingen er anbefalt til alle (234). Selve høstingen gjøres med granulocytt-stimulerende faktor (G-CSF), uten cyklofosfamid, i motsetning til ved myelomatose, fordi cyklofosfamid i slike doser ofte er dårlig tolerert ved amyloidose (240). Både under høstingen og ved stamcelleinfusjonen er det økt risiko for arytmier og ekstravasal væskelekkasje. Disse pasientene må overvåkes nøye.
Pasienter som vurderes for HMAS bør oppfylle følgende kriterier (241):
- alder <70 år
- ECOG 0-2 (funksjonsstatus)
- NYHA klasse I-II
- systolisk BT >90 mm Hg
- troponin T (TnT) <60 ng/L
- NTproBNP <5000ng/L
- eGFR >40mL/min (for pasienter som ikke er i dialyse)
- bilirubin <1,5 x øvre referanse uten tegn til syntesesvikt.
Noen pasienter som tidlig i forløpet ikke ansees å være kandidater for HMAS kan etter induksjonsbehandlingen oppnå betydelig bedring av organfunksjonen og dermed bli aktuelle for HMAS.
6.1.1.3 Samtidig myelomatose
Hos AL-amyloidosepasienter med samtidig myelomatose vises det til handlingsprogram for myelomatose (2).
6.1.1.4 Organtransplantasjoner
I en del tilfeller av AL-amyloidose lar det seg gjøre å få meget god kontroll på plasmacelleklonen. Klinisk respons kan imidlertid ta flere år (152) fordi amyloidfibrillene løser seg langsomt. Spesielt hjertesviktpasienter kan risikere å dø av sin hjertesvikt i mellomtiden. Nyresvikt kan forårsakes av amyloidavleiring og/eller av medikamentell behandling og pasienten kan bli dialysetrengende. I begge tilfeller kan organtransplantasjon være aktuelt. Nyretransplantasjon ved amyloidose beskrives nærmere i avsnitt 6.2.2. Hjertetransplantasjon kan være et alternativ i helt spesielle tilfeller. Det omfatter unge pasienter med kardiell AL-amyloidose uten komorbiditet (242) eller annen organskade fra amyloidosen. Hvert enkelt av disse tilfellene må drøftes med og mellom avdeling for blodsykdommer, hjertemedisinsk og hjertekirurgisk avdeling ved OUS Rikshospitalet.
6.1.1.5 Behandling av refraktær sykdom, og behandling av klinisk tilbakefall
Det er ingen konsensus internasjonalt om hva som defineres som behandlingskrevende residiv. Videre behandling må veies opp mot komorbiditet, alder og forventet toksisitet av behandlingen. Det er generell enighet om at behandling er indisert ved hematologisk tilbakefall og før det har tilkommet organskade. For pasienter som ikke oppnår hematologisk VGPR etter fire sykluser eller PR etter to sykluser på aktuelt regime er det som nevnt over indikasjon for å endre behandling. Det foreligger data fra både prospektive og retrospektive studier som anbefaler Daratumumab-basert behandling til pasienter med tilbakefall/progresjon (243, 244). Hos daratumumab-refraktære pasienter har både proteasombasert behandling og immunmodulerende medikamenter (IMiDs) vist å ha effekt. For ytterligere detaljer henvises til handlingsprogrammet for AL-amyloidose (1).
6.1.2 Systemisk AA-amyloidose
Behandling av AA-amyloidose er primært kontroll av den underliggende inflammatoriske sykdommen, og gjennom å redusere nivå av sirkulerende serum amyloid A (SAA). Prognosen ved AA-amyloidose er vist å være korrelert med sirkulerende nivå av SAA (54, 111) og behandlingen avhenger av den underliggende inflammatoriske sykdommen. Randomiserte studier som sammenligner ulike behandlingsalternativer ved etablert AA-amyloidose ved de ulike inflammatoriske sykdommene mangler. Behandlingsvalg styres av historiske data med pasientserier med AA-amyloidose samt effekt på grunnsykdommen vurdert fra andre parametere som for eksempel påvirkning av SAA og inflammatorisk aktivitet. Behandlingsmål er livsforlengelse og symptomdemping.
Hos de fleste pasienter som utvikler AA-amyloidose er glomerulær proteinuri (albuminuri), med eller uten nyresvikt i form av redusert GFR, det første kliniske tegn. Rutinemessig oppfølgning med måling av proteinuri og nyrefunksjon hos risikopasienter kan bidra til tidligere diagnostikk og mer effektiv kontroll av den inflammatoriske grunnsykdommen.
Moderne sykdomsmodifiserende behandling av revmatiske sykdommer har ført til nedgang i insidens av AA-amyloidose blant revmatikere (172, 245). Monoklonale antistoffer mot henholdsvis TNFα og IL-6 er i pasientserier vist å redusere så vel nivå av SAA, som kliniske symptomer og AA-avleiringer (246, 247).
Høydose kolkisin (1,5-2 mg/dag) brukes ved familiær middelhavsfeber (FMF) og er vist å redusere hyppigheten av anfall med magesmerter, hemme utvikling av amyloidose og dermed insidens av nyresvikt ved denne tilstanden (248). Antistoffer mot interleukin 1 (anakinra, canakinumab) brukes ved autoinflammatoriske syndromer som kryopyrinopatier (CAPS) og kolkisinresistent FMF (249).
I tilfeller hvor AA-amyloidose er resultat av lymfoproliferative tilstander, som ved Castlemans sykdom, er kirurgisk fjerning av årsaken til overproduksjon av SAA vist å redusere amyloidavleiring (250). I tråd med at insidensen av AA-amyloidose er betydelig redusert i befolkningsgrupper som får antimikrobiell behandling for langvarige infeksjoner som for eksempel tuberkulose, er det vist at behandling med antimikrobielle midler ved etablert AA-amyloidose som skyldes infeksjoner kan gi tilbakegang av amyloidet, f.eks. tilbakegang av nefrotisk syndrom etter behandling med tuberkulostatika hos pasienter med tuberkulose (251).
6.1.3 Systemisk ATTR-amyloidose
Inntil nylig har det ikke vært noen etablert behandling for ATTRwt utover symptomatisk behandling. De siste årene er det utviklet spesifikk terapi for ATTR-amyloidose som kan virke på både ATTRwt og ATTRv ved flere ulike mekanismer (252). Et angrepspunkt er å redusere produksjonen via RNA inhibitorer og dermed mengden sirkulerende transtyretin (se avsnitt 6.1.3.1). Et annet er å stabilisere transtyretin i sin native konformasjon og dermed hindre at det foldes galt (se avsnitt 6.1.3.2). Et tredje og mindre etablert behandlingsprinsipp er å angripe deponert amyloid med fibrill-løsende stoffer (253, 254) som doksysyklin og tauro-ursodeoksykolsyre (TUDCA), og er fortsatt å regne som eksperimentell behandling da det ikke er gjennomført randomiserte kontrollerte studier (RCT) (se avsnitt 6.3).
For den arvelige formen for transtyretinamyloidose, ATTRv, har levertransplantasjon (se avsnitt 6.1.4.1) vært brukt som en «kirurgisk genterapi», hvor jo den friske leveren vil produsere villtype-TTR i stedet for en ekstra ustabil TTR-variant. Levertransplantasjon er nå nesten forlatt som behandling for ATTRv etter at det har kommet andre alternative behandlingsformer for å redusere eller å stabilisere det fibrilledannende proteinet TTR.
6.1.3.1 TTR-syntese hemmende behandling
For både ATTRwt og ATTRv er det vist at leverens produksjon av TTR kan hemmes på gennivå med medikamenter som degraderer TTR mRNA enten via mRNA-hemning i cellekjerne eller i cytoplasma (255, 256). Small interfering RNA (siRNA) og antisense oligonukleotid (ASO) er RNA-varianter som kan blokkere syntese av spesifikke proteiner på transkripsjonsnivå, dvs. ved å hindre at transkribert mRNA blir translatert til protein. Dette er en del av kroppens regulering av genekspresjon, og kan utnyttes når man ønsker å hemme produksjonen av enkelte proteiner. Ved å inkorporere slike RNA-varianter i fettpartikler klarer man å få en relativt selektiv leveranse til leveren, og slik få størst mulig effekt ut av hver dose. Ved ATTR skal behandling med siRNA og ASO hemme mRNA i lever for å supprimere leverens transtyretin produksjon og dermed sirkulerende transtyretin i blodet.
Hovedstudiene på disse medikamentene er gjort på pasienter med ATTRv med nevropati. For patisiran (APOLLO A) ser man på sekundære kardiale endepunkter i en subgruppe som viste redusert verdier av venstre ventrikkel masse og reduksjon av NTproBNP. APOLLO B-studien som er ventet ferdig i 2022, ser på effekt av patisiran hos pasienter med kardial ATTR (255). Helios B er en RCT på kardial ATTR med vutrisiran (mot placebo) som var ferdig inkludert i august 21 med tre års oppfølging. To TTR-syntesehemmere, patisiran og inotersen er godkjent av European Medical Agency (EMA) og US Food Drug Administration (FDA) for ATTRv.
6.1.3.2 Konformasjonsstabiliserende behandling
Prinsippet for konformasjonsstabiliserende behandling er å stabilisere sirkulerende transtyretin i en tetramerstruktur; siden det er TTR-monomerer som danner fibriller. Dissosiasjon av tetrameren til monomerer er et kritisk steg for fibrilledannelse fra TTR (19, 253, 257). Ulike TTR-mutasjoner som øker sannsynligheten for amyloiddannelse er vist å destabilisere tetramerstrukturen. Konformasjons-stabilisering gjør at fibrilledannelsen blir hemmet og har effekt på både villtype og variant ATTR. Konformasjonsstabiliserende behandling med tafamidis ble først godkjent av EMA i 2011 for ATTRv med polynevropati og fra 2020 for ATTRv og ATTRwt hjerteamyloidose etter ATTR-ACT studien som viste effekt på total dødelighet og kardiovaskulær hospitalisering for pasienter i NYHA kl. I og II (ATTR-ACT-studien) (257). En «open label extension» til ATTR-ACT hvor placebogruppen ble konvertert til enten 80 mg eller 20 mg tafamidis daglig, viste en reduksjon i dødelighet og kardiovaskulær hospitalisering til fordel for 80 mg-gruppen (258). Konformasjonsstabiliserende behandling med tafamidis anbefales (klasse 1-anbefaling) for ATTR hjerteamyloidose i 2021-utgaven av European Society of Cardiology (ESC) guidelines (259). Tafamidis (Vyndaqel) er godkjent i Norge, men Beslutningsforum har ikke godkjent medikamentet for generell bruk i Helseforetakene1.
6.1.4 Andre systemiske amyloidoser
6.1.4.1 Levertransplantasjon ved arvelige amyloidoser
Antallet årlige levertransplantasjoner for arvelige amyloidoser har etter 2010 vært betydelig lavere enn tidligere fordi mange velger å være avventende når det nå finnes medikamentell behandling for ATTR-amyloidose.
Levertransplantasjon har vært førstelinjebehandling for arvelige amyloidoser når det amyloide fibrilleproteinet er produsert i leveren. Levertransplantasjon kan dermed anses som en «kirurgisk genterapi», når pasienten i stedet for en lever med en sykdomsfrembringende mutasjon får en frisk lever som produsere den normale formen for det aktuelle proteinet, f.eks. TTRwt. Totalt er det per i dag utført over 2000 levertransplantasjoner i verden for arvelige amyloidoser (260), og majoriteten av disse for arvelig ATTR-amyloidose (ATTRv). Hittil er ingen levertransplantasjoner utført for arvelig amyloidose i Norge, hvor vi har få påviste tilfeller av arvelig amyloidose. I Sverige er det utført > 150 levertransplantasjoner for arvelig amyloidose, de fleste for ATTR Val30Met, som forekommer endemisk i Sverige.
Overlevelsesdata ved levertransplantasjon er rapportert i Familial Amyloidotic World Transplant Registry. Registeret viser en 20-årsoverlevelse etter levertransplantasjon på > 50 % for pasienter med ATTRv (n=1940) (260). Dette er en betydelig forbedring fra historiske data med gjennomsnittlig overlevelse på 10 til 15 år fra symptomdebut. Overlevelse etter transplantasjon er best for dem med færrest år fra symptomdebut til transplantasjon. Etter levertransplantasjon går produksjonen av mutert protein ned, men da flere av fibrilleproteinene også kan produseres i noen grad utenfor lever, vil ikke alltid levertransplantasjon eliminere alt av det sirkulerende muterte proteinet. Dette er vist for eksempel for apoA1, som også kan dannes i tynntarm (199). For ATTRv er det vist at etter levertransplantasjon reduseres sirkulerende TTRv og sykdomsaktiviteten stopper hos de fleste, men hos noen pasienter fortsetter amyloiddeponering, nå med normal type TTR (261).
6.1.4.2. ALECT2-amyloidose
Det er ingen spesifikk terapi for ALECT2 per i dag. I det sykdommen er såpass nylig oppdaget er det også relativt sparsomt med prognostiske data. I et amerikansk materiale på 72 pasienter med påvist ALECT2 hadde en tredjedel av pasientene utviklet endestadie nyresvikt (eGFR< 15) etter en median oppfølgingstid på 26 mnd., og median nyreoverlevelse var 5 år (209). I et britisk materiale på 24 pasienter med median oppfølgingstid på 4,8 år var median estimert nyreoverlevelse fra diagnose 8,2 år (211).
6.1.4.3 Dialyserelatert amyloidose
Behandling av Aβ2M er å øke utskillelsen av β2-mikroglobulin; da opphopning av β2-mikroglobulin er en forutsetning for Aβ2M. Aβ2M sees hos pasienter som har hatt endestadie (dialysekrevende) nyresvikt i flere år. Symptomgivende Aβ2M er relativt sjelden i Norge fordi en stor andel av pasienter med endestadie nyresvikt blir nyretransplantert, og dermed får økt sin utskillelse av β2M. Det er ikke vist forskjell mellom hemodialyse og peritonealdialyse, men invers relasjon til nyrefunksjon. I de tilfeller hvor pasienten ikke kan bli nyretransplantert, tilstreber man best mulig utskillelse av β2M gjennom dialysen, ved hemodialyse teller spesielt dialysemembranenes porestørrelse og total dialyse-mengde, som innbefatter dialysetid, dialyseeffektivitet og ultrafiltrasjonsvolum. Det er vist at insidensen av karpaltunnelsyndrom blant dialysepasienter er minkende i senere år, hvilket man relaterer til bedre dialyse (216, 262).
Ved påvist Aβ2M kan det være aktuelt med støttende behandling som eksisjon av amyloide masser fra karpaltunnelen, og kirurgisk behandling av patologiske frakturer.
6.1.5 Lokaliserte amyloidoser
Lokalisert amyloidose har generelt god prognose. Ofte vil det ikke være behov for noen behandling, eventuelt lokal kirurgisk behandling eller strålebehandling av affisert område. Systemisk behandling kan være aktuelt i sjeldne tilfeller ved f.eks. obstruerende AL-deponeringer som ikke er tilgjengelige for lokal behandling, og må anses som utprøvende behandling.
6.1.5.1 Alzheimers sykdom og cerebral amyloid angiopati
Acetylkolinestereasehemmere og memantin er etablert symptombehandling av Alzheimers sykdom. Det har vært flere forsøk med amyloidrettet behandling mot Alzheimers sykdom de siste tiårene. Et monoklonalt antistoff rettet mot amyloid-[beta], aducanumab, fikk nylig en akselerert FDA godkjenning i USA (263). Dette er den første sykdomsmodifiserende behandlingen mot Alzheimers sykdom, og den første nye behandlingen siden 2003. Godkjennelsen er imidlertid kontroversiell og under debatt, grunnet usikker behandlingseffekt og en bivirkningsprofil med kortikale mikroblødinger og hevelse i hjernens hvite substans.
Det fins per i dag ingen spesifikk behandling av cerebral amyloid angiopati. CAA medfører økt blødningsrisiko, og man må være tilbakeholden med blodfortynnende behandling og avdekke og behandle eventuell hypertensjon.
6.2 Organstøttende behandling
6.2.1 Hjerte
Det er sparsom dokumentasjon på behandling av hjertesvikt ved hjerteamyloidose, og behandlingsprinsippene ligner dem man bruker ved hypertrofisk kardiomyopati. Det er likevel fundamentale forskjeller mellom myocyttsykdommen hypertrofisk kardiomyopati (HCM) og amyloid hjertesykdom som er en ekstracellulær avleiringssykdom, og som har klart dårligere prognose. Prinsippet er å finne en balanse mellom høye fyllingstrykk, slagvolum og frekvens. Amyloidavleiring i hjertet gjør ventriklene stivere, dvs. fører til et restriktivt fyllingsmønster slik at det kreves høyere fyllingstrykk for å oppnå samme slagvolum. Høye fyllingstrykk gir stuvning fra venstre ventrikkel bakover i lungekretsløpet og fra høyre ventrikkel til lever, intestinale organer og ekstremiteter. Ved reduksjon av fyllingstrykket (ved diuretika), minskes preload og dette senker slagvolumet. Pasienter med hjerteamyloidose krever tett oppfølgning da de ofte responderer forskjellig og kan være utfordrende å balansere hemodynamisk.
De fleste pasienter blir diagnostisert med hjerteamyloidose når de allerede har symptomgivende hjertesvikt og restriktivt fyllingsmønster/høye fyllingstrykk. Initial behandling er da vanndrivende legemidler som kan gi mindre stuvning i lunge- og systemkretsløpet, men må brukes med forsiktighet, da reduksjon av fyllingstrykk senker preload som gir redusert slagvolum. Dermed kan en del pasienter raskere merke effekten av redusert minuttvolum, med økende asteni, matthet og trøtthet i muskulatur. De fleste pasienter vil likevel merke symptomlindring, da dyspnø er et plagsomt symptom.
Ved svært tidlig diagnose kan betablokker ha en hjertebeskyttende effekt (264). Betablokker kan forsøkes hos pasienter som har hypertrofi, men ikke økt fyllingstrykk. Betablokker kan virke gunstig ved høy hvilepuls eller anstrengelsesutløst takykardi eller for å frekvensregulere atrieflimmer. Betablokkaden må trappes opp forsiktig, pasientene må monitoreres med tanke på AV-blokk, og følges tett, helst av kardiolog. Ved svimmelhet, synkope eller påvist AV-blokk grad I-II må betablokker seponeres og pasientene må rytmeovervåkes. Pasienter med hjerteamyloidose er ekstra følsomme for den negative inotrope og kronotrope effekten av betablokkere. Før start med betablokker må pasienten være i best mulig hemodynamisk balanse, dvs. med minst mulig stuvning og godt forhåndsbehandlet med diuretika. Betablokkereffekten måles først og fremst klinisk, men de objektive parameterne puls, blodtrykk, kreatinin, NTproBNP og EKG med måling av PR intervall kan og bør også følges. Ved manifest atrieflimmer bør betablokker være basismedikasjon ved behov for frekvenskontroll. Ved betablokkerbivirkning eller mangelfull effekt kan amiodarone brukes og betablokker trappes ned til en lavere dose, eventuelt seponeres. Flekainid, digitalis og de nyere antiarytmika er kontraindisert (265). Digitalis akkumuleres i myokard (265). Ut fra hjertesviktstudier (ikke amyloid-spesifikke) anbefales hvilepuls ved atrieflimmer å ligge på ca. 80/min og ved anstrengelse ikke å overstige 120-130/min. Pasienter med amyloidose har som regel lavt blodtrykk grunnet lavt minuttvolum, autonom dysfunksjon og forandret kartonus. Ved diastolisk dysfunksjon/HFpEF skal afterload/BT være normalt. Det er ikke lavt blodtrykk i seg selv som bestemmer grad av BT-medikasjon, men symptomer på hypotensjon eller tegn til lavt perfusjonstrykk i andre organer avgjør om man skal trekker tilbake blodtrykksmedisiner.
ATTR-amyloidose, både i arvelig og nativ form synes å respondere noe bedre på hjertemedisiner enn AL-amyloidose og symptomatisk hypotensjon kan oppstå. Hos disse pasientene kan ACE-hemmer eller AT-blokker introduseres i lave doser med langsom opptrapping hvis hypertensjon. Eldre pasienter kan ha andre årsaker enn amyloidose til hjertesykdom og bør vurderes i lys av dette.
Ved atrioventrikulære blokk eller bradykardi er pacemaker indisert etter vanlige retningslinjer. Tokammerpacemaker er som regel indisert. Pasienter som er paceavhengige kan utvikle systolisk svikt eller få redusert sitt minuttvolum ved kontinuerlig pacing i høyre ventrikkel og indikasjon for biventrikulær pacemaker vurderes etter vanlige retningslinjer. Implanterbar hjertestarter (ICD) vurderes ved ventrikulær arytmi med eller uten symptomer. Små studier har vist begrenset effekt av ICD og mindre ventrikulær arytmi enn forventet. En publisert studie viste at bradykardi med påfølgende iskemi ofte foranlediget plutselig hjertedød (108).
Antitrombotisk behandling brukes hos pasienter som har koronarsykdom eller ved symptomatisk angina pectoris selv om koronarkar er vurdert til å være normale. Patologi i mikrosirkulasjon kan forekomme og acetylsalisylsyre synes logisk å bruke på vanlig klinisk indikasjon ved angina. Ved høye fyllingstrykk og atriell blodstase og/eller intrakardiale tromber kan warfarin eller direktevirkende perorale antikoagulasjonsmidler (DOAK) være indisert også ved sinusrytme. Det er høyere slagfrekvens ved ATTR hjerteamyloidose enn ved VVH av annen årsak. Dette kan sees som spontankontrast ved ekkokardiografi. En relativ kontraindikasjon er anamnestiske blødninger tydende på økt karfragilitet og/eller økt INR som tegn på funksjonell faktor X-mangel.
Ved langtkommen hjerteamyloidose vil det etter hvert utvikles en systolisk svikt. Dette er et svært alvorlig prognostisk tegn og pasientene merker symptomforverring. Behandlingsprinsipper ved systolisk hjertesvikt (ACE- eller AT-blokker) bør om mulig forsøkes, men i svært lave doser og med nøye oppfølging. Mange pasienter vil ikke tolerere dette grunnet hypotensjon, men responsen er individuell. I endestadiet ses ofte en dilatert kardiomyopati med systolisk og diastolisk svikt. På grunn av den tilgrunnliggende pseudohypertrofien er ventrikkelveggene ofte ikke så tynne som ved dilatert kardiomyopati av andre årsaker.
Ved Rikshospitalet er hjertetransplantasjon og deretter HMAS (se avsnitt 6.1.1.2) hittil blitt utført hos én pasient med AL amyloidose. Pasienten overlevde i 4 år med god hjertefunksjon, men fikk dessverre residiv av sin grunnsykdom. Pasientserier fra USA er beskrevet med klart dårligere resultat enn hos andre hjertetransplanterte. Noen pasienter er også hjertetransplantert uten å gjennomgå HMAS. Ofte er det andre organkomplikasjoner som begrenser muligheten for hjertetransplantasjon (266).
6.2.2 Nyre
Endestadie nyresvikt er en viktig årsak til morbiditet og mortalitet ved flere former for amyloidose. Nyresviktbehandling ved amyloidose skiller seg ikke fra nyresviktbehandling ved andre årsaker til nyresvikt. Nyretransplantasjon er førstevalg som nyreerstattende behandling også ved amyloidose, men har vært kontroversielt i enkelte miljøer grunnet bekymring for redusert graft- og pasientoverlevelse med rekurrent amyloid i nyregraftet og ekstrarenal organaffeksjon av amyloid. Imidlertid viser studier både fra Norge internasjonalt at pasienter som er selektert til transplantasjon har både graft- og pasientoverlevelse i samme størrelsesorden som ved diabetes nefropati (169, 267, 268).
I et materiale fra London presenteres data for 99 pasienter som i perioden 1989-2018 er nyretransplantert på bakgrunn av enten AA-amyloidose (n=48) eller AL-amyloidose (n=51) (268). Studien viser 1-, 5- og 10-års graftoverlevelse (sensurert for død) på 94, 91 and 78 % for AA amyloidose, og 98, 93 and 93 % for AL amyloidose. Median pasientoverlevelse var henholdsvis 13,1 år for AA og 7,9 år for AL. Rekurrent amyloid i graftet ble påvist hos 9 pasienter med AA amyloidose og 7 pasienter med AL-amyloidose, men bare to pasienter mistet graftet pga. dette. Overlevelsen i amyloidosegruppen var på samme nivå som for pasienter nyretransplantert på bakgrunn av diabetes nefropati.
I Norge er pasienter med endestadie nyresvikt på bakgrunn av amyloidose nyretransplantert siden 1974 (169), de fleste av disse med AA-amyloidose, men også noen med AL-amyloidose. Blant 89 norske pasienter med AA-amyloidose som ble nyretransplantert i perioden 1998-2008 var 70 % i live etter 5 år, med 5-års graftoverlevelse sensurert for død på 88 % (269) (samt upubliserte data). Median alder på transplantasjonstidspunkt var 50 år. I løpet av oppfølgningsperioden, døde 59 av 89 pasienter, median 7,3 år etter transplantasjonen. Kardiovaskulære hendelser og infeksjoner var som for andre nyretransplanterte de hyppigste dødsårsakene. Sepsis var tre ganger hyppigere som dødsårsak hos pasientene med AA-amyloidose, og var dødsårsak hos 29 % av disse. 11 % av nyregraftene gikk tapt pga. rekurrent AA-amyloid i graftet.
6.2.3 Mage- og tarmkanalen
Behandlingen består av sykdomsspesifikk behandling og symptomreduserende tiltak. Pasientene trenger ofte kostveiledning for å redusere symptomer. Små og hyppige måltider anbefales. Ved gastroparese anbefales lavt innhold av fett og fiber samt forsiktighet med alkohol og niktotin (89). Flytende, kvernet og most mat tømmes lettere fra magesekken. Metoklopramid eller erytromycin kan være aktuell som symptomatisk behandling for å stimulere ventrikkeltømming. Dersom hovedproblemet er kvalme, kan antiemetika uten prokinetiske egenskaper benyttes, for eksempel ondansetron (89). Ved diare eller obstipasjon behandles disse på vanlig måte med tarmregulerende midler. Ved behov for næringstilførsel, kan både sondenæring og parenteral næringstilførsel vurderes. Forebygging og overvåkning for å forhindre mangeltilstander er en viktig del av behandling og oppfølging.
6.2.4 Polynevropati
Det finnes ingen spesifikk behandling for generell polynevropati. Men symptomatisk behandling for smertefull tynnfibernevropati i form av medikamenter som gabapentin eller pregabalin kan dempe smertene, men må tas regelmessig og har bivirkninger. Ved blæreparese som følge av autonom nevropati med selvkateterisering institueres. Sildenafil har særlig god effekt på ereksjonssvikt når årsaken er nevrogen. Ved nevropati er det også viktig å gjøre pasienten oppmerksom på risiko for sår som ikke gror og oppfordre til regelmessig fotstell. Ortoser kan være gode hjelpemidler ved uttalt motorisk nevropati med dropp-fot.
6.3 Andre behandlingsformer under utvikling for ulike typer amyloidose
Flere nye behandlingsformer for ulike former for amyloidose er under utvikling og vil bare bli helt kort berørt i dette avsnittet. Vi henviser til oppdatert litteratur og informasjon om pågående studier i databasen https://www.clinicaltrials.gov/. De fleste nye behandlingsformene er skreddersydd for én type amyloidose, rettet mot enten syntese av fibrilleproteinet, stabilisering av fibrilleproteinets konformasjon eller angrep på avleiret amyloid.
For ATTR er flere TTR-syntesehemmere, inkludert revusiran og vutrisiran samt modifiseringer av inotersen under utvikling (252). En pasientserie med genredigering med CRISPR teknologi er publisert for ATTR (270). Diflunisal har vært brukt off-label som konformasjonsstabilisering for ATTR (271), men i hovedsak forlatt etter godkjenning av tafamidis. Flere stabilisatorer av tetramerstruktur er under utvikling, bl.a. tolcapone og AG10 (252). Grønn-te-ekstrakt (Epigallocatechin-3-gallate (EGCG) har vært lovende i prekliniske studier med hensyn på konformasjonsstabilisering for ATTR og dessuten å kunne løse opp fibriller for AL, men har heller ikke dokumentert effekt i RCT (252).
Doksysyklin har blitt brukt i behandling av både ATTR og AL med hjerteaffeksjon, basert på forsøk i dyremodeller, kasuistikker og små pasientserier. En ny RCT på doksysyklin ved hjerteaffeksjon ved AL viste imidlertid ingen effekt (272).
Flere typer plasmacellerettet behandling, mange basert på erfaring med tilsvarende behandling ved myelomatose, er under utprøving ved AL (273). Passiv immunisering med monoklonale antistoffer rettet mot de amyloide fibrillene kan være aktuelt for alle former for amyloidose. Det monoklonale antistoffet CAEL-101 (11-1F4) er under utprøving for AL-amyloidose (274).
Behandling rettet mot ekstrafibrillære komponenter er potensielt uavhengig av type amyloidose og dermed håp for felles terapi. Lovende resultater dyremodeller og prekliniske studier har imidlertid ikke blitt reprodusert i RCT (275, 276).
1 Godkjent per juli 2022. For vilkår se: https://nyemetoder.no/metoder/tafamidis-vyndaqel-indikasjon-ii
Referanser
1. AL amyloidose handlingsprogram 2021 (sist faglig oppdatert 11. mars 2021, lest 09. november 2021); 2021. Available from: https://www.legeforeningen.no/foreningsledd/fagmed/Norsk-selskap-for-hematologi/handlingsprogram/.
2. Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av maligneblodsykdommer [nettdokument]. Oslo: Helsedirektoratet (sist faglig oppdatert 27. mai 2020, lest 09. november 2021). 2018. Available from: https://www.helsedirektoratet.no/retningslinjer/maligne-blodsykdommer-handlingsprogram.
3. Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, et al. Misdiagnosis of hereditaryamyloidosis as AL (primary) amyloidosis. NEnglJ Med. 2002;346(23):1786-91.
4. Comenzo RL, Zhou P, Fleisher M, Clark B, Teruya-Feldstein J. Seeking confidence in the diagnosis ofsystemic AL (Ig light-chain) amyloidosis: patients can have both monoclonal gammopathies andhereditary amyloid proteins. Blood. 2006;107(9):3489-91.
5. Lane T, Fontana M, Martinez-Naharro A, Quarta CC, Whelan CJ, Petrie A, et al. Natural History, Quality ofLife, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation. 2019;140(1):16-26.
6. McCausland KL, White MK, Guthrie SD, Quock T, Finkel M, Lousada I, et al. Light Chain (AL) Amyloidosis:The Journey to Diagnosis. Patient. 2018;11(2):207-16.
7. Schjesvold FH, Sjo M, Tangen JM, Hammerstrom J, Brinch L. [High-dose treatment of systemic AL-amyloidosis with autologous stem cell support]. Tidsskr Nor Laegeforen. 2008;128(12):1392-6.
8. Schjesvold FH. Utredning av amyloidose. Tidsskr Nor Laegeforen. 2013;133(3):266.
9. Cohen AS, Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverseorigins. Nature. 1959;183(4669):1202-3.
10. Bergstrom J, Murphy CL, Weiss DT, Solomon A, Sletten K, Hellman U, et al. Two different types ofamyloid deposits-apolipoprotein A-IV and transthyretin-in a patient with systemic amyloidosis. LabInvest. 2004;84(8):981-8.
11. Sidiqi MH, McPhail ED, Theis JD, Dasari S, Vrana JA, Drosou ME, et al. Two types of amyloidosispresenting in a single patient: a case series. Blood Cancer J. 2019;9(3):30.
12. Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJM, Sekijima Y, et al. Amyloid nomenclature2020: update and recommendations by the International Society of Amyloidosis (ISA) nomenclaturecommittee. Amyloid: the international journal of experimental and clinical investigation: the officialjournal of the International Society of Amyloidosis. 2020;27(4):217-22.
13. Colombat M, Barres B, Renaud C, Ribes D, Pericard S, Camus M, et al. Mass spectrometry-basedproteomic analysis of parathyroid adenomas reveals PTH as a new human hormone-derived amyloidfibril protein. Amyloid: the international journal of experimental and clinical investigation: the officialjournal of the International Society of Amyloidosis. 2021;28(3):153-7.
14. Ichimata S, Katoh N, Abe R, Yoshinaga T, Kametani F, Yazaki M, et al. A case of novel amyloidosis: glucagon-derived amyloid deposition associated with pancreatic neuroendocrine tumour. Amyloid: the international journal of experimental and clinical investigation: the official journal of the InternationalSociety of Amyloidosis. 2021;28(1):72-3.
15. Hampel H, Hardy J, Blennow K, Chen C, Perry G, Kim SH, et al. The Amyloid-beta Pathway in Alzheimer'sDisease. Mol Psychiatry. 2021: doi: 10.1038/s41380-021-01249-0. Online ahead of print.
16. Koga S, Sekiya H, Kondru N, Ross OA, Dickson DW. Neuropathology and molecular diagnosis ofSynucleinopaties. Molecular Neurodegeneration. 2021;16. 10.1186/s13024-021-00501-z
17. Tasaki M, Lavatelli F, Obici L, Obayashi K, Miyamoto T, Merlini G, et al. Age-related amyloidosis outsidethe brain: A state-of-the-art review. Ageing Res Rev. 2021;70:101388.
18. Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetesmellitus. Physiol Rev. 2011;91(3):795-826.
19. Gertz MA, Dispenzieri A. Systemic Amyloidosis Recognition, Prognosis, and Therapy: A SystematicReview. JAMA. 2020;324(1):79-89.
20. Ravichandran S, Lachmann HJ, Wechalekar AD. Epidemiologic and Survival Trends in Amyloidosis, 1987-2019. N Engl J Med. 2020;382(16):1567-8.
21. Hemminki K, Li X, Forsti A, Sundquist J, Sundquist K. Incidence and survival in non-hereditary amyloidosisin Sweden. BMC Public Health. 2012;12:974.
22. Pinney JH, Smith CJ, Taube JB, Lachmann HJ, Venner CP, Gibbs SD, et al. Systemic amyloidosis in England:an epidemiological study. British journal of haematology. 2013;161(4):525-32.
23. Staron A, Connors LH, Ruberg FL, Mendelson LM, Sanchorawala V. A new era of amyloidosis: the trendsat a major US referral centre. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2019;26(4):192-6.
24. Janssen S, van Rijswijk MH, Meijer S, Ruinen L, Van der Hem GK. Systemic amyloidosis: a clinical surveyof 144 cases. NethJMed. 1986;29(11):376-85.
25. Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O'Fallon WM, et al. Incidence and natural history ofprimary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood.1992;79(7):1817-22.
26. Kyle RA, Larson DR, Kurtin PJ, Kumar S, Cerhan JR, Therneau TM, et al. Incidence of AL Amyloidosis inOlmsted County, Minnesota, 1990 through 2015. Mayo Clinic proceedings. 2019;94(3):465-71.
27. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world studyusing US claims data. Blood Adv. 2018;2(10):1046-53.
28. Papa R, Lachmann HJ. Secondary, AA, Amyloidosis. Rheum Dis Clin North Am. 2018;44(4):585-603.
29. Manner I, Sagedal S, Roger M, Os I. Renal amyloidosis in intravenous heroin addicts with nephroticsyndrome and renal failure. ClinNephrol. 2009;72(3):224-8.
30. Cornwell GG, Murdoch WL, Kyle RA, Westermark P, Pitkanen P. Frequency and distribution of senilecardiovascular amyloid. A clinicopathologic correlation. AmJMed. 1983;75(4):618-23.
31. Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, et al. Senile systemic amyloidosisaffects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: apopulation-based autopsy study. AnnMed. 2008;40(3):232-9.
32. Castano A, Narotsky DL, Hamid N, Khalique OK, Morgenstern R, DeLuca A, et al. Unveiling transthyretincardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoingtranscatheter aortic valve replacement. European heart journal. 2017;38(38):2879-87.
33. Reisæter AV, Åsberg A. Annual report 2020. The Norwegian Renal Registry 2021. Available from: http://nephro.no/nnr/AARSRAPPORT_NNR_2020.pdf
34. Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP, et al. Emerging concepts in sporadiccerebral amyloid angiopathy. Brain. 2017;140(7):1829-50.
35. Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy Diagnosis of CardiacTransthyretin Amyloidosis. Circulation. 2016;133(24):2404-12.
36. Glenner GG. Amyloid deposits and amyloidosis. The beta-fibrilloses. NEnglJMed. 1980;302(23):1283-92.
37. Westermark GT, Fandrich M, Westermark P. AA Amyloidosis: Pathogenesis and Targeted Therapy.Annual review of pathology. 2015;10:321-44.
38. Lansbury PT, Jr. Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis anddisease. Proceedings of the National Academy of Sciences of the United States of America.1999;96(7):3342-4.
39. Husby G, Marhaug G, Dowton B, Sletten K, Sipe JD. Serum amyloid A (SAA): biochemistry, genetics andthe pathogenesis of AA amyloidosis. Amyloid:IntJExpClinInvest. 1994;1:119-37.
40. Dember LM, Jaber BL. Dialysis-related amyloidosis: late finding or hidden epidemic? SeminDial.2006;19(2):105-9.
41. Hawkins PN. Serum amyloid P component scintigraphy for diagnosis and monitoring amyloidosis.CurrOpinNephrolHypertens. 2002;11(6):649-55.
42. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. NEnglJMed. 2003;349(6):583-96.
43. Bemporad F, Chiti F. Protein misfolded oligomers: experimental approaches, mechanism of formation,and structure-toxicity relationships. Chemistry & biology. 2012;19(3):315-27.
44. Pepys MB, Booth DR, Hutchinson WL, Gallimore JR, Collins PM, Hohenester E. Amyloid P Component. Acritical review. Amyloid:IntJExpClinInvest. 1997;4:274-95.
45. Snow AD, Kisilevsky R. Temporal Relationship between Glycosaminoglycan Accumulation and AmyloidDeposition during Experimental Amyloidosis - a Histochemical-Study. Laboratory Investigation.1985;53(1):37-44.
46. Li JP, Galvis ML, Gong F, Zhang X, Zcharia E, Metzger S, et al. In vivo fragmentation of heparan sulfate byheparanase overexpression renders mice resistant to amyloid protein A amyloidosis.ProcNatlAcadSciUSA. 2005;102(18):6473-7.
47. Axelrad MA, Kisilevsky R, Willmer J, Chen SJ, Skinner M. Further characterization of amyloid-enhancingfactor. LabInvest. 1982;47(2):139-46.
48. Lundmark K, Westermark GT, Nystr"m S, Murphy CL, Solomon A, Westermark P. Transmissibility ofsystemic amyloidosis by a prion-like mechanism. ProcNatlAcadSciUSA. 2002;99(10):6979-84.
49. Jarrett JT, Lansbury PT, Jr. Amyloid fibril formation requires a chemically discriminating nucleation event:studies of an amyloidogenic sequence from the bacterial protein OsmB. Biochemistry.1992;31(49):12345-52.
50. Rochet JC, Lansbury PT, Jr. Amyloid fibrillogenesis: themes and variations. CurrOpinStructBiol.2000;10(1):60-8.
51. Hurshman AR, White JT, Powers ET, Kelly JW. Transthyretin aggregation under partially denaturingconditions is a downhill polymerization. Biochemistry. 2004;43(23):7365-81.
52. Hawkins PN, Richardson S, MacSweeney JE, King AD, Vigushin DM, Lavender JP, et al. Scintigraphicquantification and serial monitoring of human visceral amyloid deposits provide evidence for turnoverand regression. QJ Med. 1993;86(6):365-74.
53. Hawkins PN, Myers MJ, Lavender JP, Pepys MB. Diagnostic radionuclide imaging of amyloid: biologicaltargeting by circulating human serum amyloid P component. Lancet. 1988;1(8600):1413-8.
54. Gillmore JD, Lovat LB, Persey MR, Pepys MB, Hawkins PN. Amyloid load and clinical outcome in AAamyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet.2001;358(9275):24-9.
55. Lachmann HJ, Gallimore R, Gillmore JD, Carr-Smith HD, Bradwell AR, Pepys MB, et al. Outcome insystemic AL amyloidosis in relation to changes in concentration of circulating free immunoglobulin lightchains following chemotherapy. BrJ Haematol. 2003;122(1):78-84.
56. Hrncic R, Wall J, Wolfenbarger DA, Murphy CL, Schell M, Weiss DT, et al. Antibody-mediated resolutionof light chain-associated amyloid deposits. AmJPathol. 2000;157(4):1239-46.
57. Solomon A, Weiss DT, Wall JS. Therapeutic potential of chimeric amyloid-reactive monoclonal antibody11-1F4. ClinCancer Res. 2003;9(10 Pt 2):3831S-8S.
58. Tennent GA, Lovat LB, Pepys MB. Serum amyloid P component prevents proteolysis of the amyloid fibrilsof Alzheimer disease and systemic amyloidosis. ProcNatlAcadSciUSA. 1995;92(10):4299-303.
59. Westermark P, Davey E, Lindbom K, Enqvist S. Subcutaneous fat tissue for diagnosis and studies ofsystemic amyloidosis. Acta histochemica. 2006;108(3):209-13.
60. Halloush RA, Lavrovskaya E, Mody DR, Lager D, Truong L. Diagnosis and typing of systemic amyloidosis:The role of abdominal fat pad fine needle aspiration biopsy. CytoJournal. 2010;6:24.
61. Cohen OC, Sharpley F, Gilbertson JA, Wechalekar AD, Sachchithanantham S, Mahmood S, et al. The valueof screening biopsies in light-chain (AL) and transthyretin (ATTR) amyloidosis. Eur J Haematol.2020;105(3):352-6.
62. Garcia Y, Collins AB, Stone JR. Abdominal fat pad excisional biopsy for the diagnosis and typing ofsystemic amyloidosis. Human pathology. 2018;72:71-9.
63. Bogov B, Lubomirova M, Kiperova B. Biopsy of subcutaneus fatty tissue for diagnosis of systemicamyloidosis. Hippokratia. 2008;12(4):236-9.
64. Fish R, Pinney J, Jain P, Addison C, Jones C, Jayawardene S, et al. The incidence of major hemorrhagiccomplications after renal biopsies in patients with monoclonal gammopathies. Clin J Am Soc Nephrol.2010;5(11):1977-80.
65. Soares SM, Fervenza FC, Lager DJ, Gertz MA, Cosio FG, Leung N. Bleeding complications aftertranscutaneous kidney biopsy in patients with systemic amyloidosis: single-center experience in 101patients. Am J Kidney Dis. 2008;52(6):1079-83.
66. Di Giovanni B, Gustafson D, Delgado DH. Amyloid transthyretin cardiac amyloidosis: diagnosis andmanagement. Expert Rev Cardiovasc Ther. 2019;17(9):673-81.
67. Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral.Heart. 2011;97(1):75-84.
68. Picken MM, Linke RP. Nephrotic Syndrome Due to an Amyloidogenic Mutation in Fibrinogen A alphaChain. Journal of the American Society of Nephrology. 2009;20(8):1681-5.
69. Leung N, Nasr SH, Sethi S. How I treat amyloidosis: the importance of accurate diagnosis and amyloidtyping. Blood. 2012;120(16):3206-13.
70. Vrana JA, Gamez JD, Madden BJ, Theis JD, Bergen HR, 3rd, Dogan A. Classification of amyloidosis by lasermicrodissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood.2009;114(24):4957-9.
71. Brambilla F, Lavatelli F, Di Silvestre D, Valentini V, Rossi R, Palladini G, et al. Reliable typing of systemicamyloidoses through proteomic analysis of subcutaneous adipose tissue. Blood. 2012;119(8):1844-7.
72. Olsen KE, Sletten K, Westermark P. Fragments of the constant region of immunoglobulin light chains areconstituents of AL-amyloid proteins. Biochemical and biophysical research communications.1998;251(2):642-7.
73. Bodi K, Prokaeva T, Spencer B, Eberhard M, Connors LH, Seldin DC. AL-Base: a visual platform analysis tool for the study of amyloidogenic immunoglobulin light chain sequences. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2009;16(1):1-8.
74. Gilbertson JA, Theis JD, Vrana JA, Lachmann H, Wechalekar A, Whelan C, et al. A comparison of immunohistochemistry and mass spectrometry for determining the amyloid fibril protein from formalin-fixed biopsy tissue. J Clin Pathol. 2015;68(4):314-7.
75. Picken MM. Proteomics and mass spectrometry in the diagnosis of renal amyloidosis. Clin Kidney J. 2015;8(6):665-72.
76. Said SM, Sethi S, Valeri AM, Leung N, Cornell LD, Fidler ME, et al. Renal amyloidosis: origin and clinicopathologic correlations of 474 recent cases. Clin J Am Soc Nephrol. 2013;8(9):1515-23.
77. Brunger AF, Nienhuis HLA, Bijzet J, Hazenberg BPC. Causes of AA amyloidosis: a systematic review. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2020;27(1):1-12.
78. Garcia-Pavia P, Rapezzi C, Adler Y, Arad M, Basso C, Brucato A, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. European heart journal. 2021;42(16):1554-68.
79. Cariou E, Bennani Smires Y, Victor G, Robin G, Ribes D, Pascal P, et al. Diagnostic score for the detection of cardiac amyloidosis in patients with left ventricular hypertrophy and impact on prognosis. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2017;24(2):101-9.
80. Falk RH, Quarta CC. Echocardiography in cardiac amyloidosis. Heart Fail Rev. 2015;20(2):125-31.
81. Saito M, Imai M, Wake D, Higaki R, Nakao Y, Sumimoto T, et al. Semiquantitative assessment of the relative apical sparing pattern of longitudinal strain for cardiac amyloidosis identification. Echocardiography. 2020;37(9):1422-9.
82. Saito M, Nakao Y, Higaki R, Kawachi Y, Yokomoto Y, Ogimoto A, et al. Clinical significance of the relative apical sparing pattern of longitudinal strain in patients with cardiac amyloidosis European Heart Journal. 2020;41:1012.
83. Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076-84.
84. Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112(13):2047-60.
85. Gonzalez-Lopez E, Gagliardi C, Dominguez F, Quarta CC, de Haro-Del Moral FJ, Milandri A, et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. European heart journal. 2017;38(24):1895-904.
86. Feng D, Edwards WD, Oh JK, Chandrasekaran K, Grogan M, Martinez MW, et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation. 2007;116(21):2420-6.
87. Esplin BL, Gertz MA. Current trends in diagnosis and management of cardiac amyloidosis. Current problems in cardiology. 2013;38(2):53-96.
88. Nakov R, Suhr OB, Ianiro G, Kupcinskas J, Segal JP, Dumitrascu DL, et al. Recommendations for the diagnosis and management of transthyretin amyloidosis with gastrointestinal manifestations. Eur J Gastroenterol Hepatol. 2021;33(5):613-22.
89. Sangnes DA, Søfteland E, Biermann M, Gilja OH, Thordarson H, Dimcevski G. Gastroparese - årsaker, diagnostikk og behandling. Tidsskr Nor Laegeforen. 2016;136(9):822-6.
90. Kim SH, Han JK, Lee KH, Won HJ, Kim KW, Kim JS, et al. Abdominal amyloidosis: spectrum of radiological findings. Clinical radiology. 2003;58(8):610-20.
91. Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 2 of 2-Diagnostic criteria and appropriate utilization. J Nucl Cardiol. 2019;26(6):2065-123.
92. Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 1 of 2-evidence base and standardized methods of imaging. J Nucl Cardiol. 2019;26(6):2065-123.
93. Rapezzi C, Quarta CC, Guidalotti PL, Longhi S, Pettinato C, Leone O, et al. Usefulness and limitations of 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in the aetiological diagnosis of amyloidotic cardiomyopathy. European journal of nuclear medicine and molecular imaging. 2011;38(3):470-8.
94. Hutt DF, Quigley AM, Page J, Hall ML, Burniston M, Gopaul D, et al. Utility and limitations of 3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in systemic amyloidosis. Eur Heart J Cardiovasc Imaging. 2014;15(11):1289-98.
95. Quarta CC, Obici L, Guidalotti PL, Pieroni M, Longhi S, Perlini S, et al. High 99mTc-DPD myocardial uptake in a patient with apolipoprotein AI-related amyloidotic cardiomyopathy. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2013;20(1):48-51.
96. Hansson O, Lehmann S, Otto M, Zetterberg H, Lewczuk P. Advantages and disadvantages of the use of the CSF Amyloid beta (Abeta) 42/40 ratio in the diagnosis of Alzheimer's Disease. Alzheimers Res Ther. 2019;11(1):34.
97. Palmqvist S, Zetterberg H, Blennow K, Vestberg S, Andreasson U, Brooks DJ, et al. Accuracy of brain amyloid detection in clinical practice using cerebrospinal fluid beta-amyloid 42: a cross-validation study against amyloid positron emission tomography. JAMA Neurol. 2014;71(10):1282-9.
98. Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):186-93.
99. Fontana M, Chung R, Hawkins PN, Moon JC. Cardiovascular magnetic resonance for amyloidosis. Heart Fail Rev. 2015;20(2):133-44.
100. Mohty D, Damy T, Cosnay P, Echahidi N, Casset-Senon D, Virot P, et al. Cardiac amyloidosis: updates in diagnosis and management. Archives of cardiovascular diseases. 2013;106(10):528-40.
101. Linn J, Halpin A, Demaerel P, Ruhland J, Giese AD, Dichgans M, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology. 2010;74(17):1346-50.
102. Pinto MV, Dyck PJB, Liewluck T. Neuromuscular amyloidosis: Unmasking the master of disguise. Muscle Nerve. 2021;64(1):23-36.
103. Donnelly JP, Hanna M, Sperry BW, Seitz WH, Jr. Carpal Tunnel Syndrome: A Potential Early, Red-Flag Sign of Amyloidosis. J Hand Surg Am. 2019;44(10):868-76.
104. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020;136(23):2620-7.
105. Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18-22 April 2004. American journal of hematology. 2005;79(4):319-28.
106. Fitzgerald BT, Scalia GM, Cain PA, Garcia MJ, Thomas JD. Left atrial size--another differentiator for cardiac amyloidosis. Heart Lung Circ. 2011;20(9):574-8.
107. Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29(14):1924-33.
108. Sayed RH, Rogers D, Khan F, Wechalekar AD, Lachmann HJ, Fontana M, et al. A study of implanted cardiac rhythm recorders in advanced cardiac AL amyloidosis. European heart journal. 2014.
109. Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart: a comprehensive review. ArchInternMed. 2006;166(17):1805-13.
110. Rocken C, Peters B, Juenemann G, Saeger W, Klein HU, Huth C, et al. Atrial amyloidosis: an arrhythmogenic substrate for persistent atrial fibrillation. Circulation. 2002;106(16):2091-7.
111. Lachmann HJ, Goodman HJ, Gilbertson JA, Gallimore JR, Sabin CA, Gillmore JD, et al. Natural history and outcome in systemic AA amyloidosis. NEnglJ Med. 2007;356(23):2361-71.
112. Joss N, McLaughlin K, Simpson K, Boulton-Jones JM. Presentation, survival and prognostic markers in AA amyloidosis. QJM. 2000;93(8):535-42.
113. Verine J, Mourad N, Desseaux K, Vanhille P, Noel LH, Beaufils H, et al. Clinical and histological characteristics of renal AA amyloidosis: a retrospective study of 68 cases with a special interest to amyloid-associated inflammatory response. Human pathology. 2007;38(12):1798-809.
114. Sethi S, Theis JD. Pathology and diagnosis of renal non-AL amyloidosis. J Nephrol. 2018;31(3):343-50.
115. Yang Z, Laird A, Monaghan A, Seywright M, Ahmad I, Leung HY. Incidental seminal vesicle amyloidosis observed in diagnostic prostate biopsies--are routine investigations for systemic amyloidosis warranted? Asian journal of andrology. 2013;15(1):149-51.
116. Zhou F, Lee P, Zhou M, Melamed J, Deng FM. Primary localized amyloidosis of the urinary tract frequently mimics neoplasia: a clinicopathologic analysis of 11 cases. American journal of clinical and experimental urology. 2014;2(1):71-5.
117.Monge M, Chauveau D, Cordonnier C, Noel LH, Presne C, Makdassi R, et al. Localized amyloidosis of thegenitourinary tract: report of 5 new cases and review of the literature. Medicine (Baltimore).2011;90(3):212-22.
118.Obici L, Suhr OB. Diagnosis and treatment of gastrointestinal dysfunction in hereditary TTR amyloidosis.Clin Auton Res. 2019;29(Suppl 1):55-63.
119.Freudenthaler S, Hegenbart U, Schonland S, Behrens HM, Kruger S, Rocken C. Amyloid in biopsies of thegastrointestinal tract-a retrospective observational study on 542 patients. Virchows Arch.2016;468(5):569-77.
120.Menke DM, Kyle RA, Fleming CR, Wolfe JT, 3rd, Kurtin PJ, Oldenburg WA. Symptomatic gastricamyloidosis in patients with primary systemic amyloidosis. Mayo Clinic proceedings. 1993;68(8):763-7.
121.Cowan AJ, Skinner M, Seldin DC, Berk JL, Lichtenstein DR, O'Hara CJ, et al. Amyloidosis of thegastrointestinal tract: a 13-year, single-center, referral experience. Haematologica. 2013;98(1):141-6.
122.Wixner J, Mundayat R, Karayal ON, Anan I, Karling P, Suhr OB, et al. THAOS: gastrointestinalmanifestations of transthyretin amyloidosis - common complications of a rare disease. Orphanet J RareDis. 2014;9:61.
123.Wixner J, Suhr OB, Anan I. Management of gastrointestinal complications in hereditary transthyretinamyloidosis: a single-center experience over 40 years. Expert Rev Gastroenterol Hepatol. 2018;12(1):73-81.
124.Chang HS, Myung SJ, Yang SK, Jung HY, Lee GH, Hong WS, et al. Massive small bowel bleeding in apatient with amyloidosis. GastrointestEndosc. 2004;59(1):126-9.
125.Hazenberg BP, van Rijswijk MH. Clinical and therapeutic aspects of AA amyloidosis. BaillieresClinRheumatol. 1994;8(3):661-90.
126.Kyle RA, Greipp PR. Amyloidosis (AL). Clinical and laboratory features in 229 cases. Mayo Clinicproceedings. 1983;58(10):665-83.
127.Buck FS, Koss MN. Hepatic amyloidosis: morphologic differences between systemic AL and AA types.Human pathology. 1991;22(9):904-7.
128.Syed U, Ching Companioni RA, Alkhawam H, Walfish A. Amyloidosis of the gastrointestinal tract and theliver: clinical context, diagnosis and management. Eur J Gastroenterol Hepatol. 2016;28(10):1109-21.
129.Levine RA. Amyloid disease of the liver. Correlation of clinical, functional and morphologic features inforty-seven patients. Am J Med. 1962;33:349-57.
130.Adams D, Ando Y, Beirao JM, Coelho T, Gertz MA, Gillmore JD, et al. Expert consensus recommendationsto improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268(6):2109-22.
131.Gertz MA, Lacy MQ, Dispenzieri A. Amyloidosis. Hematology/oncology clinics of North America.1999;13(6):1211-33, ix.
132.Adams D, Lozeron P, Theaudin M, Denier C, Fagniez O, Rerat K, et al. Varied patterns of inaugural light-chain (AL) amyloid polyneuropathy: a monocentric study of 24 patients. Amyloid: the internationaljournal of experimental and clinical investigation: the official journal of the International Society ofAmyloidosis. 2011;18 Suppl 1:98-100.
133.Tracy JA, Dyck PJ, Dyck PJ. Primary amyloidosis presenting as upper limb multiple mononeuropathies.Muscle Nerve. 2010;41(5):710-5.
134.Luigetti M, Papacci M, Bartoletti S, Marcaccio A, Romano A, Sabatelli M. AL amyloid neuropathymimicking a chronic inflammatory demyelinating polyneuropathy. Amyloid: the international journal ofexperimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2012;19(1):53-5.
135.Berk JL, O'Regan A, Skinner M. Pulmonary and tracheobronchial amyloidosis. SeminRespirCrit Care Med. 2002;23(2):155-65.
136.Pitkanen P, Westermark P, Cornwell GG, 3rd. Senile systemic amyloidosis. Am J Pathol. 1984;117(3):391-9.
137.Bach-Gansmo T, Wien TN, Londalen A, Halvorsen E. Myocardial uptake of bone scintigraphic agentsassociated with increased pulmonary uptake. Clin Physiol Funct Imaging. 2016;36(3):237-41.
138.Lachmann HJ, Hawkins PN. Amyloidosis and the lung. Chron Respir Dis. 2006;3(4):203-14.
139.Baqir M, Kluka EM, Aubry MC, Hartman TE, Yi ES, Bauer PR, et al. Amyloid-associated cystic lung diseasein primary Sjogren's syndrome. Respir Med. 2013;107(4):616-21.
140.Willander H, Askarieh G, Landreh M, Westermark P, Nordling K, Keranen H, et al. High-resolutionstructure of a BRICHOS domain and its implications for anti-amyloid chaperone activity on lungsurfactant protein C. Proceedings of the National Academy of Sciences of the United States of America.2012;109(7):2325-9.
141. Fernandez-Flores A. Cutaneous amyloidosis: a concept review. Am J Dermatopathol. 2012;34(1):1-14; quiz 5-7.
142. Woollons A, Black MM. Nodular localized primary cutaneous amyloidosis: a long-term follow-up study. Br J Dermatol. 2001;145(1):105-9.
143. D'Souza A, Theis JD, Vrana JA, Dogan A. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2014;21(2):71-5.
144. Kay J. Beta2-microglobulin amyloidosis. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 1997;4:187-211.
145. Dammacco R, Merlini G, Lisch W, Kivela TT, Giancipoli E, Vacca A, et al. Amyloidosis and Ocular Involvement: an Overview. Semin Ophthalmol. 2020;35(1):7-26.
146. Kang S, Dehabadi MH, Rose GE, Verity DH, Amin S, Das-Bhaumik R. Ocular amyloid: adnexal and systemic involvement. Orbit. 2020;39(1):13-7.
147. Schmidt EK, Mustonen T, Kiuru-Enari S, Kivela TT, Atula S. Finnish gelsolin amyloidosis causes significant disease burden but does not affect survival: FIN-GAR phase II study. Orphanet J Rare Dis. 2020;15(1):19.
148. Harris G, Lachmann H, Hawkins P, Sandhu G. One Hundred Cases of Localized Laryngeal Amyloidosis - Evidence for Future Management. Laryngoscope. 2021.
149. Said SM, Reynolds C, Jimenez RE, Chen B, Vrana JA, Theis JD, et al. Amyloidosis of the breast: predominantly AL type and over half have concurrent breast hematologic disorders. Mod Pathol. 2013;26(2):232-8.
150. Gillmore JD, Hawkins PN. Pathophysiology and treatment of systemic amyloidosis. NatRevNephrol. 2013;9(10):574-86.
151. Yamada M, Naiki H. Cerebral amyloid angiopathy. Progress in molecular biology and translational science. 2012;107:41-78.
152. Dispenzieri A, Gertz MA, Buadi F. What do I need to know about immunoglobulin light chain (AL) amyloidosis? Blood Rev. 2012;26(4):137-54.
153. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-48.
154. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30(9):989-95.
155. Turesson I, Velez R, Kristinsson SY, Landgren O. Patterns of multiple myeloma during the past 5 decades: stable incidence rates for all age groups in the population but rapidly changing age distribution in the clinic. Mayo Clinic proceedings. 2010;85(3):225-30.
156. Rajkumar SV, Gertz MA, Kyle RA. Primary systemic amyloidosis with delayed progression to multiple myeloma. Cancer. 1998;82(8):1501-5.
157. Li J, Huang Z, Duan MH, Zhang W, Chen M, Cao XX, et al. Characterization of immunoglobulin lambda light chain variable region (IGLV) gene and its relationship with clinical features in patients with POEMS syndrome. Annals of hematology. 2012;91(8):1251-5.
158. Li J, Zhou DB, Huang Z, Jiao L, Duan MH, Zhang W, et al. Clinical characteristics and long-term outcome of patients with POEMS syndrome in China. Annals of hematology. 2011;90(7):819-26.
159. Dispenzieri A, Kyle RA, Lacy MQ, Rajkumar SV, Therneau TM, Larson DR, et al. POEMS syndrome: definitions and long-term outcome. Blood. 2003;101(7):2496-506.
160. Kuwabara S, Dispenzieri A, Arimura K, Misawa S, Nakaseko C. Treatment for POEMS (polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes) syndrome. The Cochrane database of systematic reviews. 2012;6:CD006828.
161. Dispenzieri A. POEMS Syndrome: 2019 Update on diagnosis, risk-stratification, and management. American journal of hematology. 2019;94(7):812-27.
162. Randall RE, Williamson WC, Jr., Mullinax F, Tung MY, Still WJ. Manifestations of systemic light chain deposition. Am J Med. 1976;60(2):293-9.
163. Benditt EP, Eriksen N. Chemical classes of amyloid substance. AmJ Pathol. 1971;65(1):231-52.
164. Kisilevsky R, Manley PN. Acute-phase serum amyloid A: perspectives on its physiological and pathological roles. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2012;19(1):5-14.
165. Yamada T. Serum amyloid A (SAA): a concise review of biology, assay methods and clinical usefulness. ClinChemLabMed. 1999;37(4):381-8.
166. Maury CP. Comparative study of serum amyloid A protein and C-reactive protein in disease. Clin Sci (Lond). 1985;68(2):233-8.
167. Cunnane G, Grehan S, Geoghegan S, McCormack C, Shields D, Whitehead AS, et al. Serum amyloid A in the assessment of early inflammatory arthritis. JRheumatol. 2000;27(1):58-63.
168. Falck HM, Maury CP, Teppo AM, Wegelius O. Correlation of persistently high serum amyloid A protein and C-reactive protein concentrations with rapid progression of secondary amyloidosis. BrMedJ (ClinResEd). 1983;286(6375):1391-3.
169. Hartmann A, Holdaas H, Fauchald P, Nordal KP, Berg KJ, Talseth T, et al. Fifteen years' experience with renal transplantation in systemic amyloidosis. TransplInt. 1992;5(1):15-8.
170. Myllykangas-Luosujarvi R, Aho K, Kautiainen H, Hakala M. Amyloidosis in a nationwide series of 1666 subjects with rheumatoid arthritis who died during 1989 in Finland. Rheumatology(Oxford). 1999;38(6):499-503.
171. Laiho K, Tiitinen S, Kaarela K, Helin H, Isomaki H. Secondary amyloidosis has decreased in patients with inflammatory joint disease in Finland. ClinRheumatol. 1999;18(2):122-3.
172. Immonen K, Finne P, Gronhagen-Riska C, Pettersson T, Klaukka T, Kautiainen H, et al. A marked decline in the incidence of renal replacement therapy for amyloidosis associated with inflammatory rheumatic diseases - data from nationwide registries in Finland. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2011;18(1):25-8.
173. Lane T, Pinney JH, Gilbertson JA, Hutt DF, Rowczenio DM, Mahmood S, et al. Changing epidemiology of AA amyloidosis: clinical observations over 25 years at a single national referral centre. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2017;24(3):162-6.
174. Lane T, Loeffler JM, Rowczenio DM, Gilbertson JA, Bybee A, Russell TL, et al. AA amyloidosis complicating the hereditary periodic fever syndromes. Arthritis Rheum. 2013;65(4):1116-21.
175. Ensari C, Ensari A, Tumer N, Ertug E. Clinicopathological and epidemiological analysis of amyloidosis in Turkish patients. NephrolDialTransplant. 2005;20(8):1721-5.
176. Abdallah E, Waked E. Incidence and clinical outcome of renal amyloidosis: a retrospective study. SaudiJKidney DisTranspl. 2013;24(5):950-8.
177. Blank N, Hegenbart U, Dietrich S, Brune M, Beimler J, Rocken C, et al. Obesity is a significant susceptibility factor for idiopathic AA amyloidosis. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2018;25(1):37-45.
178. Stankovic Stojanovic K, Georgin-Lavialle S, Poitou C, Buob D, Amselem S, Grateau G, et al. AA amyloidosis is an emerging cause of nephropathy in obese patients. Eur J Intern Med. 2017;39:e18-e20.
179. Uzum G, Kaya FO, Uzum AK, Kucukyilmaz M, Gunes ME, Duzkoylu Y, et al. Amyloid goiter associated with amyloidosis secondary to rheumatoid arthritis. Case reports in medicine.2013:792413.
180. Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, et al. Senile Systemic Amyloidosis: Clinical Features at Presentation and Outcome. J Am Heart Assoc. 2013;2(2):e000098.
181. Westermark P, Bergstrom J, Solomon A, Murphy C, Sletten K. Transthyretin-derived senile systemic amyloidosis: clinicopathologic and structural considerations. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2003;10 Suppl 1:48-54.
182. Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. JAmHeart Assoc. 2013;2(2):e000098.
183. Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, Prognosis, and Therapy of Transthyretin Amyloidosis. J Am Coll Cardiol. 2015;66(21):2451-66.
184. Holmgren G, Costa PM, Andersson C, Asplund K, Steen L, Beckman L, et al. Geographical distribution of TTR met30 carriers in northern Sweden: discrepancy between carrier frequency and prevalence rate. J Med Genet. 1994;31(5):351-4.
185. Olsson M, Jonasson J, Cederquist K, Suhr OB. Frequency of the transthyretin Val30Met mutation in the northern Swedish population. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2014;21(1):18-20.
186. Drugge U. Familial amyloidosis with polyneuropathy (FAP) in Sweden. Some notes on its historilcal background. In: Costa PP, de Freitas, A.F., Saraiva M.J.M., editor. Familial amyloidotic polyneuropathy and other transthyretin related disorders. 3. Porto. Portugal: Arquivos de Medicina; 1990. p. 13-5.
187. Zeldenrust SR. Genotype--phenotype correlation in FAP. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2012;19 Suppl 1:22-4.
188. Yamashita T, Hamidi Asl K, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile122 allele frequency in an African-American population. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2005;12(2):127-30.
189. Quarta CC, Buxbaum JN, Shah AM, Falk RH, Claggett B, Kitzman DW, et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372(1):21-9.
190. Ranlov I, Alves IL, Ranlov PJ, Husby G, Costa PP, Saraiva MJ. A Danish kindred with familial amyloid cardiomyopathy revisited: identification of a mutant transthyretin-methionine111 variant in serum from patients and carriers. Am J Med. 1992;93(1):3-8.
191. Jacobson DR, McFarlin DE, Kane I, Buxbaum JN. Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet. 1992;89(3):353-6.
192. Lobato L, Rocha A. Transthyretin amyloidosis and the kidney. Clin J Am Soc Nephrol. 2012;7(8):1337-46.
193. Rowczenio DM, Noor I, Gillmore JD, Lachmann HJ, Whelan C, Hawkins PN, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35(9):E2403-12.
194. Benson MD, Liepnieks JJ, Kluve-Beckerman B. Hereditary systemic immunoglobulin light-chain amyloidosis. Blood. 2015;125(21):3281-6.
195. Valleix S, Gillmore JD, Bridoux F, Mangione PP, Dogan A, Nedelec B, et al. Hereditary systemic amyloidosis due to Asp76Asn variant beta2-microglobulin. N Engl J Med. 2012;366(24):2276-83.
196. Gillmore JD, Lachmann HJ, Rowczenio D, Gilbertson JA, Zeng CH, Liu ZH, et al. Diagnosis, pathogenesis, treatment, and prognosis of hereditary fibrinogen A alpha-chain amyloidosis. J AmSocNephrol. 2009;20(2):444-51.
197. Rowczenio D, Stensland M, de Souza GA, Strøm EH, Gilbertson JA, Taylor G, et al. Renal amyloidosis associated with five novel variants in the fibrinogen A alpha chain protein. Kidney International Reports. 2017;2:461-9.
198. Sethi S, Theis JD, Shiller SM, Nast CC, Harrison D, Rennke HG, et al. Medullary amyloidosis associated with apolipoprotein A-IV deposition. Kidney Int. 2012;81(2):201-6.
199. Rowczenio D, Dogan A, Theis JD, Vrana JA, Lachmann HJ, Wechalekar AD, et al. Amyloidogenicity and clinical phenotype associated with five novel mutations in apolipoprotein A-I. Am J Pathol. 2011;179(4):1978-87.
200. Benson MD, Liepnieks JJ, Yazaki M, Yamashita T, Hamidi AK, Guenther B, et al. A new human hereditary amyloidosis: the result of a stop-codon mutation in the apolipoprotein AII gene. Genomics. 2001;72(3):272-7.
201. Nasr SH, Dasari S, Hasadsri L, Theis JD, Vrana JA, Gertz MA, et al. Novel Type of Renal Amyloidosis Derived from Apolipoprotein-CII. Journal of the American Society of Nephrology: JASN. 2017;28(2):439-45.
202. Valleix S, Verona G, Jourde-Chiche N, Nedelec B, Mangione PP, Bridoux F, et al. D25V apolipoprotein C-III variant causes dominant hereditary systemic amyloidosis and confers cardiovascular protective lipoprotein profile. Nat Commun. 2016;7:10353.
203. Maury CP. Gelsolin-related amyloidosis. Identification of the amyloid protein in Finnish hereditary amyloidosis as a fragment of variant gelsolin. J Clin Invest. 1991;87(4):1195-9.
204. Solomon JP, Page LJ, Balch WE, Kelly JW. Gelsolin amyloidosis: genetics, biochemistry, pathology and possible strategies for therapeutic intervention. Crit Rev Biochem Mol Biol. 2012;47(3):282-96.
205. Pihlamaa T, Suominen S, Kiuru-Enari S. Familial amyloidotic polyneuropathy type IV--gelsolin amyloidosis. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2012;19 Suppl 1:30-3.
206. Sethi S, Theis JD, Quint P, Maierhofer W, Kurtin PJ, Dogan A, et al. Renal amyloidosis associated with a novel sequence variant of gelsolin. Am J Kidney Dis. 2013;61(1):161-6.
207. Efebera YA, Sturm A, Baack EC, Hofmeister CC, Satoskar A, Nadasdy T, et al. Novel gelsolin variant as the cause of nephrotic syndrome and renal amyloidosis in a large kindred. Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 2014;21(2):110-2.
208. Picken MM. Alect2 amyloidosis: primum non nocere (first, do no harm). Kidney Int. 2014;86(2):229-32.
209. Said SM, Sethi S, Valeri AM, Chang A, Nast CC, Krahl L, et al. Characterization and outcomes of renal leukocyte chemotactic factor 2-associated amyloidosis. 2014(1523-1755 (Electronic)).
210. Larsen CP, Kossmann RJ, Beggs ML, Solomon A, Walker PD. Clinical, morphologic, and genetic features of renal leukocyte chemotactic factor 2 amyloidosis. Kidney Int. 2014;86:378-82.
211. Rezk T, Gilbertson JA, Rowczenio D, Bass P, Lachmann HJ, Wechalekar AD, et al. Diagnosis, pathogenesis and outcome in leucocyte chemotactic factor 2 (ALECT2) amyloidosis. Nephrol Dial Transplant. 2018;33(2):241-7.
212. Benson MD, James S, Scott K, Liepnieks JJ, Kluve-Beckerman B. Leukocyte chemotactic factor 2: A novel renal amyloid protein. Kidney Int. 2008;74(2):218-22.
213. Murphy CL, Wang S, Kestler D, Larsen C, Benson D, Weiss DT, et al. Leukocyte chemotactic factor 2 (LECT2)-associated renal amyloidosis: a case series. AmJKidney Dis. 2010;56(6):1100-7.
214. Yamamoto S, Gejyo F. Historical background and clinical treatment of dialysis-related amyloidosis. BiochimBiophysActa. 2005;1753(1):4-10.
215. Jadoul M, Garbar C, Noel H, Sennesael J, Vanholder R, Bernaert P, et al. Histological prevalence of beta 2-microglobulin amyloidosis in hemodialysis: a prospective post-mortem study. Kidney Int. 1997;51(6):1928-32.
216. Kaneko S, Yamagata K. Hemodialysis-related amyloidosis: Is it still relevant? Semin Dial. 2018;31(6):612-8.
217. Stoppini M, Bellotti V. Systemic amyloidosis: lessons from beta2-microglobulin. J Biol Chem. 2015;290(16):9951-8.
218. Haggqvist B, Naslund J, Sletten K, Westermark GT, Mucchiano G, Tjernberg LO, et al. Medin: an integral fragment of aortic smooth muscle cell-produced lactadherin forms the most common human amyloid. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(15):8669-74.
219. Olsen KE, Sletten K, Sandgren O, Olsson H, Myrvold K, Westermark P. What is the role of giant cells in AL-amyloidosis? Amyloid: the international journal of experimental and clinical investigation: the official journal of the International Society of Amyloidosis. 1999;6(2):89-97.
220. Merrimen JL, Alkhudair WK, Gupta R. Localized amyloidosis of the urinary tract: case series of nine patients. Urology. 2006;67(5):904-9.
221. Raja K, Ahmed E, Mubarak M, Iqbal T, Hassan SM. Primary localized amyloidosis of urinary bladder: a case report and review of literature. Nephro-urology monthly. 2013;5(5):994-6.
222. Al Hussain H, Edward DP. Anterior orbit and adnexal amyloidosis. Middle East African journal of ophthalmology. 2013;20(3):193-7.
223. D'Souza A, Theis J, Quint P, Kyle R, Gertz M, Zeldenrust S, et al. Exploring the amyloid proteome in immunoglobulin-derived lymph node amyloidosis using laser microdissection/tandem mass spectrometry. American journal of hematology. 2013;88(7):577-80.
224. Cordier JF. Pulmonary amyloidosis in hematological disorders. Seminars in respiratory and critical care medicine. 2005;26(5):502-13.
225. Tabatabai G, Baehring J, Hochberg FH. Primary amyloidoma of the brain parenchyma. Arch Neurol. 2005;62(3):477-80.
226. Westermark P. Localized AL amyloidosis: a suicidal neoplasm? Upsala journal of medical sciences. 2012;117(2):244-50.
227. Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015;313(19):1924-38.
228. Roberts RO, Aakre JA, Kremers WK, Vassilaki M, Knopman DS, Mielke MM, et al. Prevalence and Outcomes of Amyloid Positivity Among Persons Without Dementia in a Longitudinal, Population-Based Setting. JAMA Neurol. 2018;75(8):970-9.
229. Charidimou A, Friedrich JO, Greenberg SM, Viswanathan A. Core cerebrospinal fluid biomarker profile in cerebral amyloid angiopathy: A meta-analysis. Neurology. 2018;90(9):e754-e62.
230. Muchtar E, Gertz MA, Kumar SK, Lacy MQ, Dingli D, Buadi FK, et al. Improved outcomes for newly diagnosed AL amyloidosis between 2000 and 2014: cracking the glass ceiling of early death. Blood. 2017;129(15):2111-9.
231. Muchtar E, Gertz MA, Lacy MQ, Go RS, Buadi FK, Dingli D, et al. Ten-year survivors in AL amyloidosis: characteristics and treatment pattern. British journal of haematology. 2019;187(5):588-94.
232. Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, Therneau TM, et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2004;22(18):3751-7.
233. Kastritis E, Palladini G, Minnema MC, Wechalekar AD, Jaccard A, Lee HC, et al. Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N Engl J Med. 2021;385(1):46-58.
234. Muchtar E, Dispenzieri A, Gertz MA, Kumar SK, Buadi FK, Leung N, et al. Treatment of AL Amyloidosis: Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) Consensus Statement 2020 Update. Mayo Clinic proceedings. 2021;96(6):1546-77.
235. Jaccard A, Moreau P, LeBlond V, Leleu X, Benboubker L, Hermine O, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. NEnglJ Med. 2007;357(11):1083-93.
236. Cibeira MT, Sanchorawala V, Seldin DC, Quillen K, Berk JL, Dember LM, et al. Outcome of AL amyloidosis after high-dose melphalan and autologous stem cell transplantation: long-term results in a series of 421 patients. Blood. 2011;118(16):4346-52.
237. Sharpley FA, Petrie A, Mahmood S, Sachchithanantham S, Lachmann HJ, Gillmore JD, et al. A 24-year experience of autologous stem cell transplantation for light chain amyloidosis patients in the United Kingdom. British journal of haematology. 2019;187(5):642-52.
238. Gutierrez-Garcia G, Cibeira MT, Rovira M, Fernandez de Larrea C, Tovar N, Rodriguez-Lobato LG, et al. Improving security of autologous hematopoietic stem cell transplant in patients with light-chain amyloidosis. Bone marrow transplantation. 2019;54(8):1295-303.
239. Tandon N, Muchtar E, Sidana S, Dispenzieri A, Lacy MQ, Dingli D, et al. Revisiting conditioning dose in newly diagnosed light chain amyloidosis undergoing frontline autologous stem cell transplant: impact on response and survival. Bone marrow transplantation. 2017;52(8):1126-32.
240. Devine SM. Re-examining the role of high-dose chemotherapy in the treatment of light chain amyloidosis. Biology of blood and marrow transplantation: journal of the American Society for Blood and Marrow Transplantation. 2014;20(1):14-9.
241. Gertz MA, Lacy MQ, Dispenzieri A, Kumar SK, Dingli D, Leung N, et al. Refinement in patient selection to reduce treatment-related mortality from autologous stem cell transplantation in amyloidosis. Bone marrow transplantation. 2013;48(4):557-61.
242. Kamble RT. Orthotopic heart transplant facilitated autologous hematopoietic stem cell transplantation in light-chain amyloidosis. Methodist DeBakey cardiovascular journal. 2012;8(3):17-8.
243. Roussel M, Merlini G, Chevret S, Arnulf B, Stoppa AM, Perrot A, et al. A prospective phase 2 trial of daratumumab in patients with previously treated systemic light-chain amyloidosis. Blood. 2020;135(18):1531-40.
244. Kimmich CR, Terzer T, Benner A, Dittrich T, Veelken K, Carpinteiro A, et al. Daratumumab for systemic AL amyloidosis: prognostic factors and adverse outcome with nephrotic-range albuminuria. Blood. 2020;135(18):1517-30.
245. Hazenberg BP, van Rijswijk MH. Where has secondary amyloid gone? AnnRheumDis. 2000;59(8):577-9.
246. Okuda Y, Ohnishi M, Matoba K, Jouyama K, Yamada A, Sawada N, et al. Comparison of the clinical utility of tocilizumab and anti-TNF therapy in AA amyloidosis complicating rheumatic diseases. Modern rheumatology / the Japan Rheumatism Association. 2014;24(1):137-43.
247. Pukitis A, Zake T, Groma V, Ostrovskis E, Skuja S, Pokrotnieks J. Effect of infliximab induction therapy on secondary systemic amyloidosis associated with Crohn's disease: case report and review of the literature. J Gastrointestin Liver Dis. 2013;22(3):333-6.
248. Zemer D, Pras M, Sohar E, Modan M, Cabili S, Gafni J. Colchicine in the prevention and treatment of the amyloidosis of familial Mediterranean fever. NEnglJ Med. 1986;314(16):1001-5.
249. Ter Haar N, Lachmann H, Ozen S, Woo P, Uziel Y, Modesto C, et al. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Annals of the rheumatic diseases. 2013;72(5):678-85.
250. Lachmann HJ, Gilbertson JA, Gillmore JD, Hawkins PN, Pepys MB. Unicentric Castleman's disease complicated by systemic AA amyloidosis: a curable disease. QJM: monthly journal of the Association of Physicians. 2002;95(4):211-8.
251. Castellano I, Gomez-Martino JR, Hernandez MT, Novillo R, Covarsi A. [Remission of nephrotic syndrome caused by renal amyloidosis secondary to pulmonary tuberculosis after tuberculostatic treatment]. Nefrologia: publicacion oficial de la Sociedad Espanola Nefrologia. 2001;21(1):88-91.
252. Dohrn MF, Ihne S, Hegenbart U, Medina J, Zuchner SL, Coelho T, et al. Targeting transthyretin - Mechanism-based treatment approaches and future perspectives in hereditary amyloidosis. J Neurochem. 2021;156(6):802-18.
253. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872-91.
254. Marques N, Azevedo O, Almeida AR, Bento D, Cruz I, Correia E, et al. Specific Therapy for Transthyretin Cardiac Amyloidosis: A Systematic Literature Review and Evidence-Based Recommendations. J Am Heart Assoc. 2020;9(19):e016614.
255. Adams D, Gonzalez-Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):11-21.
256. Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):22-31.
257. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018;379:1007-16.
258. Damy T, Garcia-Pavia P, Hanna M, Judge DP, Merlini G, Gundapaneni B, et al. Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and long-term extension study. Eur J Heart Fail. 2020.
259. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Bohm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. European heart journal. 2021;42(36):3599-726.
260. Ericzon BG, Wilczek HE, Larsson M, Wijayatunga P, Stangou A, Pena JR, et al. Liver Transplantation for Hereditary Transthyretin Amyloidosis: After 20 Years Still the Best Therapeutic Alternative? Transplantation. 2015;99(9):1847-54.
261. Yazaki M, Mitsuhashi S, Tokuda T, Kametani F, Takei YI, Koyama J, et al. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2007;7(1):235-42.
262. Hoshino J, Yamagata K, Nishi S, Nakai S, Masakane I, Iseki K, et al. Significance of the decreased risk of dialysis-related amyloidosis now proven by results from Japanese nationwide surveys in 1998 and 2010. Nephrol Dial Transplant. 2016;31(4):595-602.
263. FDA Grants Accelerated Approval for Alzheimer’s Drug: U.S. Food & Drug Administration; 2021 [updated 07. Jun 2021. Available from: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug.
264. Bernstein D, Fajardo G, Zhao M, Urashima T, Powers J, Berry G, et al. Differential cardioprotective/cardiotoxic effects mediated by beta-adrenergic receptor subtypes. Am J Physiol Heart Circ Physiol. 2005;289(6):H2441-9.
265. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2016.
266. Varr BC, Liedtke M, Arai S, Lafayette RA, Schrier SL, Witteles RM. Heart transplantation and cardiac amyloidosis: approach to screening and novel management strategies. The Journal of heart and lung transplantation: the official publication of the International Society for Heart Transplantation. 2012;31(3):325-31.
267. Sawinski D, Lim MA, Cohen JB, Locke JE, Weiss B, Hogan JJ, et al. Patient and Kidney Allograft Survival in Recipients With End-Stage Renal Disease From Amyloidosis. Transplantation. 2018;102(2):300-9.
268. Law S, Cohen O, Lachmann HJ, Rezk T, Gilbertson JA, Rowczenio D, et al. Renal transplant outcomes in amyloidosis. Nephrol Dial Transplant. 2021;36(2):355-65.
269. Wien TN, Leivestad T, Reisæter AV, Foss A, Hartmann A. Kidney transplantation in AA amyloidosis. Amyloid: JProtein Folding Disord. 2010;17(Suppl 1):117-.
270. Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N Engl J Med. 2021;385(6):493-502.
271. Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658-67.
272. Shen KN, Fu WJ, Wu Y, Dong YJ, Huang ZX, Wei YQ, et al. Doxycycline Combined With Bortezomib-Cyclophosphamide-Dexamethasone Chemotherapy for Newly Diagnosed Cardiac Light-Chain Amyloidosis: A Multicenter Randomized Controlled Trial. Circulation. 2021.
273. Sidiqi MH, Gertz MA. Immunoglobulin light chain amyloidosis diagnosis and treatment algorithm 2021. Blood Cancer J. 2021;11(5):90.
274. Edwards CV, Rao N, Bhutani D, Mapara MY, Radhakrishnan J, Shames S, et al. Phase 1a/b Study of Monoclonal Antibody CAEL-101 (11-1F4) in Patients with AL Amyloidosis. Blood. 2021.
275. Dember LM, Hawkins PN, Hazenberg BP, Gorevic PD, Merlini G, Butrimiene I, et al. Eprodisate for the treatment of renal disease in AA amyloidosis. NEnglJ Med. 2007;356(23):2349-60.
276. Richards DB, Cookson LM, Berges AC, Barton SV, Lane T, Ritter JM, et al. Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component. N Engl J Med. 2015;373(12):1106-14.