









Generell veileder i pediatri
10. Nyre, urinveier og kjønnsorganer
10.5 Tubulopatier
Sist faglig oppdatert: 13.04.2024
Anna Bjerre, Claus Klingenberg og Eivind Sirnes
Bakgrunn
- Tubulopatier omfatter en svært sammensatt gruppe av sjeldne sykdommer.
- Primære og sekundære, isolerte og sammensatte defekter.
- Mange av de medfødte tubulopatiene debuterer før 1 års alder.
- I nyrene blir blodet først filtrert i glomeruli og deretter skjer det en reabsorpsjon i tubuli før det endelig blir urin.
- Kunnskap om i hvilken del av tubuli de ulike reabsorpsjonsprosesser forekommer kan være med på å identifisere hvilken form for tubulopati som foreligger, se Tabell 1.
- Reabsorpsjon i proksimale tubuli er en energikrevende prosess, og ved sykdommer med nedsatt energiproduksjon (f.eks. mitokondriesykdom) kan dette medføre en tubulopati med økt utskillelse av stoffer som skulle vært reabsorbert i proksimale tubuli.
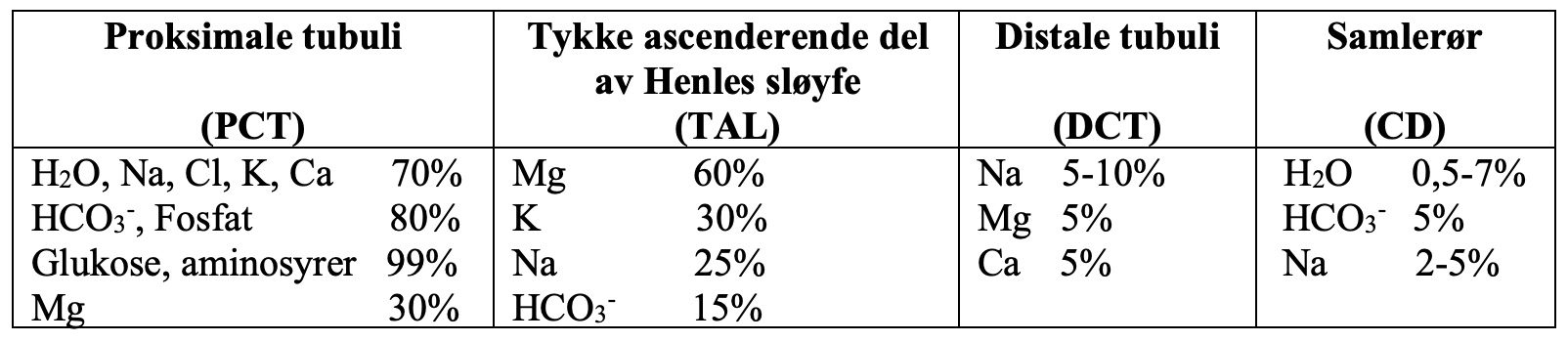
Tabell 1. Skjematisk oversikt over hvor i tubuli det foregår reabsorpsjon av ulike stoffer, og anslagvis prosent av total absorpsjon.
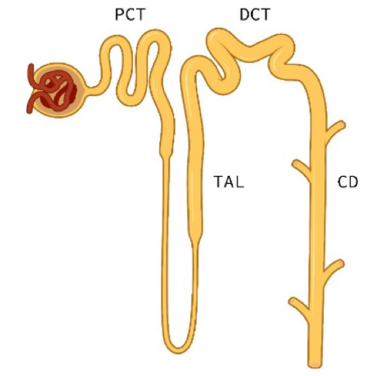
Figur 1. Tubulus systemet inndelt i proksimale tubuli (PCT), tykke ascenderende del av Henles sløyfe (TAL), distale tubuli (DCT) og samlerørene (CD).
Symptomer og funn
- Mistanke om en tubulopati bør reises ved ulike symptomer og funn som polyuri, polydipsi, uklar metabolsk acidose, uklar metabolsk alkalose, elektrolyttforstyrrelser, «faltering growth», nefrokalsinose, rakitt og funn ved urin stiks som glukosuri (med normalt blodsukker) og lettgradig proteinuri.
- Ekstrarenal affeksjon av syn, hørsel og kognitiv funksjon forekommer ved noen former.
- Tubulopatier er en svært heterogen gruppe sykdommer. Etter «Diagnostikk og utredning» omtales separat en del ulike former for tubulopatier.
- Det er viktig å være klar over at det til dels kan det være klinisk overlapp mellom noen former/typer, og terminologien kan variere noe i litteraturen.
Diagnostikk og utredning
Ved mistanke om tubulopati, skal følgende utredning utføres.
Det er gunstig å diskutere med erfaren barnelege/nefrolog.
- Blodprøver: Na, K, Cl, Ca, Mg, fosfat, PTH, syre-base status, kreatinin, urea, osmolalitet, alkalisk fosfatase, urat, TSH, fritt T3, fritt T4, albumin og 25-OH-vitamin D
- Vurder renin, aldosteron og 1,25-OH vitamin D
- Urinprøver: Stiks, pH, Na, K, Cl, Ca, osmolalitet, fosfat.
- Proteinuri beregnes med protein-kreatinin ratio.
- Kalsium-utskillelse beregnes med kalsium-kreatinin ratio.
- Aminosyrer i urin (metabolsk screening) gir informasjon om ev. aminoaciduri.
- Beta 2 mikroglobulin er forhøyet ved ev. tubulusaffeksjon.
- Fraksjonell utskillelse av ulike elektrolytter kan til tider være nyttig: (Ux/Px) x (P krea/U krea) x 100.
- Ultralyd nyrer tas med henblikk på mulig nefrokalsinose og ev. misdannelser.
- Drikke og tisselister med tanke på polyuri og omfanget av væskeinntak.
- Genetikk: Mange av disse sykdommen kan diagnostiseres ved hjelp av genetiske prøver. OUS har et genpanel for nyresykdommer som inkluderer mulighet for analyse av renal tubulær sykdom.
- Annet: Videre utredning og spesifikk diagnose kan/må deretter være mere målrettet med spesifikke undersøkelser inkl. røntgen, øyelege undersøkelse, hørselstest, nevrologisk undersøkelse etc.
Enkelte former for tubulopatier omtales nærmere
Renal tubulær acidose (RTA)
Acidose defineres som fall i pH < 7,35. Nyrenes rolle i syre-base balansen er å holde på HCO3- og skille ut H+-ioner. Tre hovedmekanismer foreligger for å regulere kroppens pH:
- Reabsorbsjon av HCO3-
- Produksjon av NH4+ i proksimale tubuli
- Sekresjon av H+-ioner i distale tubuli
Det kan altså foreligge en proksimal eller distal RTA. Diagnosen RTA kan være vanskelig å stille og først må andre årsaker til metabolsk acidose utelukkes (laktacidose, diabetes ketoacidose, nyresvikt etc.). Stikkord for å finne diagnosen er serum anion gap og urin anion gap. Det avgjørende er om det er tap av bikarbonat eller en akkumulasjon av syrer. Hos barn med persisterende hyperkloremisk acidose og normalt serum anion gap skal RTA mistenkes. Følgende skjema kan bidra i diagnostikken.
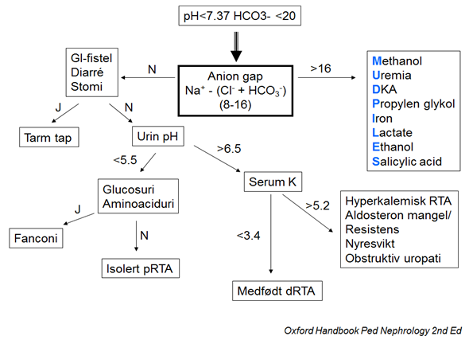
Ved en proksimal RTA (pRTA) foreligger det oftest også Fanconi syndrom (se lenger ned). Isolert pRTA er meget sjeldent. Felles symptombilde er dårlig trivsel, kvalme, oppkast og nedsatt muskelkraft.
En distal RTA (dRTA) oppstår pga av manglende evne til surgjøring i samlerørene. Urin pH er derfor > 6,5. Årsakene kan være både medfødte (genetiske) og ervervede.
Hvis man har samtidig hyperkalemi og hyponatremi (hyperkalemisk dRTA) kan årsaker være:
- Hypoaldosteronisme og aldosteronresistens (pseudohypoaldosteronisme, se senere)
- Revmatiske sykdommer (f.eks Sjøgren og SLE)
- Tubulo-interstitielle sykdommer inkl. medullær svampnyre
- Medikamenter (ACE-hemmer, ciklosporin, amfotericin B, spironolakton, topiramat etc.)
Behandling av RTA er å behandle den underliggende årsaken, samt gi ekstra tilførsel av natriumbikarbonat 1–3 mmol/kg/dag og kaliumsitrat ved nefrokalsinose.
Transient pseudohypoaldosteronisme
Alvorlig hyponatremi og hyperkalemi hos spedbarn kan skyldes tubulær aldosteronresistens. Denne tilstanden ses hos noen barn med medfødte urologiske misdannelser og blir ofte feiltolket som kongenitt binyrebarkhyperplasi (CAH). Årsaken til denne aldosteronresistensen er ukjent, men ofte ser man denne tilstanden i forbindelse med at barn med urologiske misdannelser også har en urinveisinfeksjon eller hos barn med «faltering growth». Tilstanden ses overveiende hos gutter. En ultralyd urinveier vil avdekke at det foreligger misdannelser og videre urologisk utredning gjøres etter dette. Hvis man måler aldosteron i serum vil nivåene være skyhøye. Fraksjonell Na-utskillelse er høy.
Behandlingen er rikelig med væske, bikarbonat- og NaCl tilskudd.
Vanligvis er tilstanden reversert i løpet av 1–3 måneder.
Primær pseudohypoaldosteronisme
Pseudohypoaldosteronisme type 1 (PHA1) er en svært sjelden tilstand (1:80 000).
- Renal type (PHA1B). Autosomalt dominant arv. Renalt salttap/tubulær aldosteronresistens. Symptomer avtar ofte/blir mildere tidlig i barndommen.
- Generalisert type (PHA1A). Autosomalt recessiv arv. Ved denne typen er det også salttap fra svettekjertler, spyttkjertler og tykktarm. Denne formen blir vanligvis ikke bedre.
Ved PHA1 ses alvorlig hyponatremi, hyperkalemi og manglende vektøkning/vekst samt dehydrering i spedbarnalderen. Det er typisk høy Na-utskillelse i urin. Det er ofte også metabolsk acidose som kan medføre kvalme/oppkast og slapphet. Symptomer minner om de man kan finne ved lave aldosteronverdier (aldosteronmangel/CAH), men aldosteronverdiene er høye pga. aldosteronresistens i affisert vev. For oppfølging/behandling vises det til spesiallitteratur.
En ervervet form for PHA ses som komplikasjon til takrolimus behandling.
Fanconi syndrom
Fanconi syndrom skyldes skader/medfødte defekter i reabsorpsjon i proksimale tubuli. Dette kan gi forskjellige symptomer som primær renal metabolsk acidose (pRTA), glukosuri, lettgradig proteinuri (aminoaciduri), hypofosfatemi pga. fosfaturi, rakitt og nefrokalsinose.
Det finnes mange medfødte årsaker til Fanconi syndrom som blant annet: cystinose, Lowe syndrom, mitokondriesykdommer, Wilsons sykdom, galaktosemi og hereditær fruktoseintoleranse.
Nefropatisk (infantil) cystinose er den vanligste årsaken til Fanconi syndrom. Rask diagnostikk er avgjørende for å hindre progressiv nyresvikt og for bedre tilveksten. Diagnosen stilles ved å påvise intraleukocytær cystein (prøvetaking avtales med Avdeling for medisinsk biokjemi, OUS-RH, tel. 23071049, alternativt 23071053), eller ved påvisning av krystaller i cornea ved spaltelampe og ved genetisk testing. OUS-Rikshospitalet bes kontaktet for å legge opp videre behandling, kontroller og øyelege undersøkelser.
Sekundære årsaker til Fanconi syndrom er langt vanligere. Det er en kjent komplikasjon til medikamenter (spesielt cytostatika cisplatin, ifosfamid, azatioprin), men kan også ses og etter behandling med gentamicin, ranitidin m.fl.
Bartters syndrom og Gitelman syndrom
Dette er to former for medfødte tubulopatier som begge har autosomal recessiv arvegang. Begge tilstander gir tubulær hypokalemi samt man kan se såkalt hypokalemisk-hypokloremisk metabolsk alkalose med forhøyet renin. Det kan være vanskelig å skille de to fra hverandre, selv om det er vanligere med tidlig debut hos pasienter med klassisk Bartters syndrom.
Til dels kan man skille Bartters syndrom fra Gitelman syndrom på bakgrunn av elektrolyttavvik i urin (se tabell), men genetisk diagnostikk av renal tubulær sykdom vil vanligvis avklare endelig om det er Bartters syndrom (SLC12A1, KCNJ1, CLCNKB, BSND mutasjoner, alle autosomalt recessive eller MAGED2 mutasjon som er X-bundet) eller Gitelman syndrom (SLC12A3 mutasjoner). Diskuter ev. med genetiker.
- Bartters syndrom skyldes transportdefekter svarende til den tykke ascenderende del av Henles sløyfe og er delt inn i type I–V avhengig av den genetiske defekten, der type I, II, IV ofte har symptomer fra nyfødtperioden.
- Gitelman syndrom skyldes transportdefekter av den distale del av Henles sløyfe. Symptomer oppstår ofte først senere i barne- og ungdomsalder.
| Bartter syndrom | Gitelman syndrom | |
| Start | Nyfødt/spedbarn | Barn/voksen |
| Polyhydramnion | + | - |
| Polyuri | + | - |
| Dehydrering | (+) | - |
| Salt krav | + | - |
| Dårlig tilvekst | + | -(+) |
| Muskelsvakhet | (+) | + |
| Nefrokalsinose | (+) | - |
| Hypomagnesemi | (+) | + |
| Urin NaCl | ↑ | N↑ |
| Urin Ca | N↑ | ↓ |
| Urin kons | - | N |
Behandling av Bartters syndrom: Fri væsketilførsel.
Farmakologisk hemming av prostaglandin dannelse er den primære behandlingen. Følgende NSAIDs kan brukes: Ibuprofen 15–30 mg/kg/dag delt i 3 doser ELLER celekoksib 2–10 mg/kg/dag delt i 2 doser ELLER indometacin 1–4 mg/kg/dag delt i 2–3 doser.
Ev. kalium tilskudd. Spironolakton kan brukes, men obs. hypotensjon.
Behandling av Gitelman syndrom: Primært er det tilstrekkelig med oral tilførsel av magnesium og kalium. Ev. spironolakton eller amilorid, men obs. hypotensjon. Vanligvis kan man ikke forvente at elektrolytter normaliseres helt, og man må akseptere lave verdier.
Primær nefrogen diabetes insipidus (pNDI)
- Diabetes insipidus kan være sentral (manglende ADH) eller perifer (nefrogen) med manglende respons på ADH. Sentral DI omtales i eget avsnitt.
- Rundt 90 % av tilfellene med primær NDI (pNDI) skyldes en defekt i det genet som koder for antidiuretisk hormon (ADH) V2-reseptoren (AVPR2) beliggende i celleveggen inn mot interstitiet. Denne tilstand er X-bundet.
- Kvinnelige bærer kan ha lettere symptomer og trenger av og til behandling.
- Det finnes også en dominant og en recessiv type som begge skyldes en mutasjon i genet som koder for akvaporin som er beliggende på lumen siden av cellen (AQPR2).
- 80 % av alt filtrert vann reabsorberes i proksimale tubuli og i nedadstigende del av Henles sløyfe. ADH er ansvarlig for resten av absorpsjonen ved å åpne for den passive transporten av vann i samlerørene. Denne transporten er avhengig av den osmotiske gradienten i tubuli og det omgivende interstitium.
Symptomer/funn: Symptomene ved pNDI kommer vanligvis i løpet av de første leveukene. Oppkast, dårlig trivsel, polydipsi, polyuri og dehydrering. Enkelte har en mild fenotype, en partiell ADH-resistens, og da særlig ved en defekt i akvaporin-genet kan tilstanden avdekkes først etter 1–2 år.
Diagnostikk/utredning
- Hvis s-Na > 147 mmol/l, s-osmolalitet > 300 mmol/l og u-osmolalitet < 200 mmol/l og GFR er normal, foreligger det sannsynligvis diabetes insipidus.
- En kort tørstetest (maks 6 timer) med urinsamling hvert 30.–60. min, nøye kontroll av vekt, s-Na og -osmolalitet og manglende stigning i urin-osmolalitet styrker mistanken.
- Normal eller forhøyet verdi av copeptin (gjenspeiler ADH produksjon) og/eller ev. en DDAVP-test med manglede stigning i urin-osmolalitet vil bekrefte diagnosen nefrogen diabetes insipidus.
- I dag gjør man ved sterk mistanke direkte genetisk testing som bekrefter diagnosen.
Behandling: Alltid rikelig tilgang på væske! Man bør redusere den osmotiske load med å redusere protein og salt-tilførsel.
- Hydroklortiazid 1–2 mg/kg/døgn (ev. med kalium tilskudd ved hypokalemi).
- NSAIDs kan brukes: Ibuprofen 15–30 mg/kg/dag delt i 3 doser ELLER celekoksib 2–10 mg/kg/dag delt i 2 doser ELLER indometacin 1–4 mg/kg/dag delt i 2–3 doser.
Sekundær nefrogen diabetes insipidus (sNDI)
Sekundær nefrogen diabetes insipidus (sNDI) ses spesielt ved medfødte urinveismisdannelser, nephronopthtisis, ved interstitiell nefritt, andre tubulære sykdommer som Bartter syndrom og cystinose. Det kan foreligge en betydelig polyuri som det må kompenseres for, for å unngå hypernatremisk dehydrering.
Referanser
- Kermond R, et al. A clinical approach to tubulopathies in children and young adults. Ped Nephrol 2023; 38: 651-62.
- Rees L, et al. (eds). Paediatric Nephrology 3rdedn 2019. Oxford Specialist Handbooks in Paediatrics. Chapter 6 & 7, p 119-200.
- Ashton EJ, et al. Simultaneous sequencing of 37 genes identified causative mutations in the majority of children with renal tubulopathies. Kidney Int 2018; 93: 961-7.
- Bochenhauer D, et al. Pathophysiology, diagnosis and management in nephrogenic diabetes insipidus. Nat Rev Nephrol 2015; 11: 576-88.
- Konrad et al. Diagnosis and management of Bartter syndrome: a consensus report. KI 2021; 99: 324-35.
- Veys KR, et al. Nephropatic cystinosis; an update. Curr Opin Pediatr 2017; 29: 168-78.
- Klingenberg C, et al. Transient pseudohypoaldosteronisme hos spedbarn med vesikoureteral refluks. Tidsskr Nor Legeforen 2006; 126: 315-7.
- https://www.uptodate.com med følgende temaer «Clinical manifestations and causes of nephrogenic diabetes insipidus”, “Treatment of nephrogenic diabetes insipidus”, “Bartter and Gitelman-syndromes”, “Cystinosis” og “Etiology and clinical manifestations of renal tubular acidosis in infants and children”.
- Pseudohypoaldosteronism type 1. Genetics Home Reference. https://ghr.nlm.nih.gov/condition/pseudohypoaldosteronism-type-1#statistics
- Refardt J, et al. Copeptin-based diagnosis of diabetes insipidus. Swiss Med Wkly. 2020; 150: w20237.
Tidligere versjoner
Versjon 2006: Hans-Jacob Bangstad
Revidert versjon 2009: Hans-Jacob Bangstad
Revidert versjon 2017: Anna Bjerre, Claus Klingenberg