









Generell veileder i pediatri
9.2.5 Hemoglobinsykdommer
9.2.5.1 Handlingsprogram for barn med thalassemi
Kapitteloversikt
- 9.2.1 Jernmangelanemi
- 9.2.2 Megaloblastanemeier
- 9.2.3 Hemolytiske anemier
- 9.2.4 Svikt i produksjon av røde blodlegemer
- 9.2.5 Hemoglobinsykdommer
- 9.2.5.1 Handlingsprogram for barn med thalassemi
- 9.2.5.2 Handlingsprogram for barn med sigdcellesykdom
Se behandlingsskjema for thalassemipasienter.
Innledning
Thalassemi er en gruppe genetiske sykdommer som rammer hemoglobinsyntesen, og som manifesterer seg ved redusert eller opphevet syntese av en eller flere av hemoglobinets globinkjeder. Thalassemimutasjoner er på verdensbasis svært utbredt, og thalassemiene er derfor blant de hyppigste monogene sykdommer i verden. Utbredelsen av hemoglobinsykdommer skyldes sannsynligvis en naturlig seleksjon hos befolkninger i områder der malaria er eller har vært svært utbredt, da heterozygote bærere til en viss grad er beskyttet mot malaria.
Hemoglobinmolekylet er en tetramer som består av 4 globinkjeder. Sammensetningen av kjedene varierer i løpet av utviklingen fra tidlig fostertilværelse til det endelige voksne hemoglobinet. I fosterlivet er hovedhemoglobinet HbF (føtalt hemoglobin) som består av to α- og to γ-kjeder (α2γ2). Ved fødselen utgjør HbF ca. 80 % av barnets hemoglobin. Etter fødselen skjer det en gradvis overgang til adult hemoglobin, HbA, som består av to α- og to β-kjeder (α2β2). En liten del av hemoglobinet hos voksne (ca. 2–3 %) består av HbA2, der β-kjedene er erstattet av δ-kjeder (α2δ2).
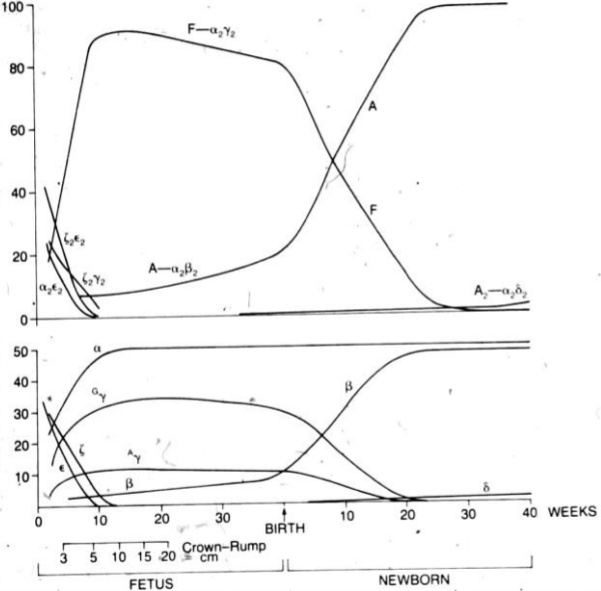
Figuren viser utviklingen gjennom fosterlivet av produksjonen av de ulike hemoglobintyper (øverst) og de tilhørende globinkjedene (nederst) på de forskjellige utviklingsstadiene. De første ukene av fosterlivet har vi Hb Portland, Hb Gower I og Hb Gower II bestående av embryonale globinkjeder. (Fra Nathan & Oski’s Hematology of Infancy and Childhood, 5th edition)
Ved α- eller β-thalassemi er det mutasjoner i ett eller flere av genene som koder for henholdsvis α- eller β-kjeder, mutasjoner som medfører redusert eller opphevet syntese av en av kjedetypene. Dette resulterer i en ubalanse mellom α- og β-kjeder, ved α-thalassemi overskudd av β-kjeder og ved β-thalassemi overskudd av α-kjeder. Det er denne ubalansen som er årsaken til de fleste kliniske manifestasjonene ved thalassemi.
HbE er en strukturell hemoglobinvariant som skyldes en punktmutasjon i genet for βglobinkjeder. Mutasjonen medfører en enkelt aminosyresubstitusjon som resulterer i en endret βkjede – βE (Bain 2006). På verdensbasis er HbE den hyppigst forekommende strukturelle Hbvariant, med en genfrekvens helt opp mot 70–80 % i enkelte områder i Sydøst-Asia. HbE er i seg selv uskyldig, også i homozygot form, men βE-kjeden produseres i redusert mengde, og i kombinasjon med β-thalassemimutasjoner blir resultatet thalassemi av varierende alvorlighetsgrad, oftest thalassemia intermedia. Andre hemoglobinvarianter som gir en thalassemilignende fenotype, er Hb Lepore (rammer δβ-kjedeproduksjonen) og Hb Constant Spring (strukturell α-kjedevariant).
α-thalassemi
Produksjonen av α-kjeder styres av 4 gener som er lokalisert to og to på kromosom nummer 16. Mutasjoner i genene for produksjon av α-kjeder er som regel delesjoner, eventuelt mutasjoner som deaktiverer ett eller flere av α-genene. Alvorlighetsgraden av α-thalassemi avhenger av hvor mange gener som er utslokket.
| Normal | α+ heterozygot | α+ homozygot | α0 heterozygot | α0/α+ – HbH-sykdom (β4) | α0 homozygot – Hb Barts (γ4) | |
Antall fungerende α-gener | 4 | 3 | 2 | 2 | 1 | 0 |
| Genotype | αα/αα | -α/αα | -α/-α | --/αα | --/-α | --/-- |
| α/β ratio | 1.0 | 0.8 | 0.6 | 0.6 | 0.3 | 0 |
| Fenotype | Normal | Normal, evt. lett mikrocytose | Mikrocytose, normal til lett nedsatt Hb | Mikrocytose, normal til lett nedsatt Hb | Som regel moderat mikrocytær, hemolytisk anemi, splenomegali | Hydrops foetalis, som regel intrauterin fosterdød |
HbH er en tetramer av β-kjeder som dannes pga. overskuddet av β-kjeder. HbH felles ut som inklusjoner i erytrocyttene etter hvert som de eldes, og medfører hemolyse. Hb Barts er en tetramer av γ-kjeder som dannes i fosterlivet hos homozygote fostre pga. en komplett mangel på α-kjeder. Hb Barts er også til stede hos nyfødte med HbH-sykdom. Hb Barts kan ikke avgi oksygen, og fostre med homozygot α0-thalassemi blir derfor alvorlig hypoksiske, utvikler hjertesvikt og hydrops og dør som regel intrauterint.
Til forskjell fra β-thalassemi som er karakterisert av benmargsekspansjon forårsaket av en aktiv ineffektiv erythropoiese, er dette ikke et vanlig funn ved α-thalassemi. α-thalassemi foråsaket av Hb Constant Spring-mutasjonen har vanligvis en alvorligere fenotype enn α-thalassemi forårsaket av delesjoner.
α+-mutasjoner er utbredt i Afrika, middelhavsområdet, Midt-Østen, deler av India og SydøstAsia, mens α0-mutasjoner er vanligst i Sydøst-Asia og øyene i Middelhavet. α+-mutasjoner har en bærerfrekvens helt oppe i 40–80 % i enkelte befolkningsgrupper.
Diagnostikk
Mikrocytose med eller uten anemi uten holdepunkter for jernmangel indiserer hemoglobinopatiutredning med henblikk på thalassemi. Hemoglobinopatiutredning består av hemoglobintyping ved elektroforese eller HPLC1 supplert med mutasjonsanalyser. Ved de milde formene av α-thalassemi (1–2 muterte gener) er hemoglobintypingen normal, men diagnosen bekreftes ved mutasjonanalysen. HbH og Hb Barts kan begge påvises ved Hb-typing, men nedbrytningsprodukter av normalt Hb kan gi en liten topp ved HPLC på samme sted som HbH.
Behandling av α-thalassemi
Milde former for α-thalassemi: Ingen behandling. Unngå unødvendig jernbehandling. Ved α0- mutasjoner kan det være risiko for alvorlig α-thalassemi hos avkom. Det bør derfor gis genetisk veiledning, gjøres testing av partner og eventuelt prenataldiagnostikk.
HbH-sykdom: Folsyretilskudd, ca. 1 mg daglig. Sporadiske transfusjoner ved behov. Unngå unødvendig jernbehandling – økt jernabsorpsjon er vanlig og kan føre til klinisk betydningsfullt jernoverskudd. Splenektomi har vanligvis liten effekt, og pga. økt trombosetendens etter splenektomi hos disse pasientene anbefales det ikke hvis det ikke er klare tegn på hypersplenisme.
Hb Barts disease: Etter intrauterine transfusjoner har enkelte barn med homozygot α0-thalassemi overlevd. Disse behandles med transfusjoner/jernkelerende behandling som ved alvorlig βthalassemi (se nedenfor). Hb Barts hydrops foetalis er assosiert med en høy frekvens av svangerskapskomplikasjoner.
β-thalassemi
Genet for koding av β-kjeder er lokalisert sammen med genene for γ- og δ-kjeder i en gruppe på kromosom nummer 11. Mutasjoner i β-genene er oftere strukturelle mutasjoner enn delesjoner.
Det er identifisert over 200 forskjellige β-thalassemimutasjoner. De kan være av alle alvorlighetsgrader og kan kode for alt fra lett nedsatt produksjon (β+-mutasjoner) til helt opphevet produksjon av β-kjeder (β0-mutasjoner). Heterozygot tilstand er vanligvis symptomfri, mens homozygot eller sammensatt (dobbelt) heterozygot tilstand gir moderat til alvorlig, transfusjonskrevende β-thalassemi. Siden HbF ikke inneholder β-kjeder, er barn med alvorlig βthalassemi friske ved fødselen, og de kliniske manifestasjonene utvikler seg gradvis etter hvert som HbF-produksjonen avtar.
Ved alvorlig β-thalassemi produseres det et stort overskudd av α-kjeder som ikke finner noen ”partner”-globinkjede å binde seg til. Frie α-kjeder kan ikke danne tetramerer. De er ustabile, presipiterer og binder seg til cellemembranen. De fleste erythrocyttforstadier med overskudd av α-kjeder destrueres derfor i benmargen – ineffektiv erythropoiese. De erythrocyttene som når perifer sirkulasjon, destrueres raskt, spesielt i milten. Anemien gir en uttalt stimulering av 1 HPLC – high pressure liquid chromatography/høytrykksvæskekromatografi erythropoiesen med resulterende ekspansjon av den erythropoietiske benmargen, noe som gir sekundære skjelettforandringer med fortynning av corticalis, deformering (spesielt i hode- og ansiktsskjelett) og frakturtendens. Det kan også dannes foci med ekstramedullær erythropoiese, som tumores av erythropoietisk vev, oftest lokalisert paravertebralt. Disse kan sees på ordinære røntgenbilder, eventuelt supplert med MR-undersøkelse. Ineffektiv erythropoiese medfører økt jernabsorpsjon som særlig er av klinisk betydning hos pasienter som ikke transfunderes regelmessig.
Alvorlighetsgraden av β-thalassemi kan modifiseres av eventuelle samtidige mutasjoner for αthalassemi som reduserer ubalansen mellom globinkjedene og derved gir et mildere klinisk forløp, eller av hereditær persistens av føtalt hemoglobin (HPFH) som også gir mindre grad av ineffektiv erythropoiese.
Diagnostikk
- Full blodstatus med retikulocytter og blodutstryk
- Jernstatus
- Hemolyseparametre (LD, haptoglobin, bilirubin, DAT)
- Hemoglobintyping ved elektroforese eller HPLC
- Mutasjonsanalyser/gensekvensering
Hb-typingen suppleres nå rutinemessig med mutasjonsanalyser, dels for å karakterisere βmutasjonen, men også med tanke på modifiserende α-mutasjoner og eventuelt HPFH. Benmargsundersøkelse er vanligvis ikke nødvendig.
Karakteristisk ved alle grader av β-thalassemi er mikrocytose, og i blodutstryket ser man hypokromasi og target cells, ved de mer alvorlige formene også uttalt grad av poikilocytose med hypokrome makrocytter (ser ut som tomme skall), bisarre mikrocytter, erythrocyttfragmenter og til dels rikelig med kjerneholdige røde blodlegemer. Retikulocyttnivået er som regel bare lett til moderat forhøyet, evt. normalt. Hemoglobintyping viser forhøyet nivå av HbA2 og/eller HbF. Ved enkelte svært milde β-thalassemimutasjoner kan HbA2 være i normalt eller tilnærmet normalt nivå, og dette vil også være tilfelle ved en eventuell samtidig mutasjon i genet for δkjeder (δβ-thalassemi). Ved homozygot/dobbelt heterozygot β0-thalassemi kan det ikke påvises HbA i det hele tatt, kun HbF og varierende mengde HbA2.
På diagnosetidspunktet kan man ikke alltid forutsi sykdommens alvorlighetsgrad selv om det ikke påvises HbA. Barnet må derfor følges nøye den første tiden for å vurdere eventuelt transfusjonsbehov. Gendiagnostikk kan i noen grad bidra til å forutsi det kliniske forløpet, men slett ikke alltid.
Klinikk
β-thalassemia major (TM)
Dette er den mest alvorlige formen for β-thalassemi og skyldes homozygoti eller dobbel heterozygoti for en alvorlig (β0-) mutasjon i genene som koder for β-kjeder. Produksjonen av βkjeder er opphevet, og barnet utvikler gradvis en alvorlig, transfusjonskrevende anemi i løpet av første eller eventuelt annet leveår. Tidspunktet for når barnet blir alvorlig sykt, bestemmes først og fremst av nivået på HbF som fortsetter å være hovedhemoglobinet i tiden postnatalt. Det er viktig å være klar over at betegnelsen «thalassemia major» innebærer transfusjonsavhengighet og er altså en klinisk definisjon. Betegnelsen «thalassemia major» er i ferd med å bli erstattet av betegnelsen transfusjonsavhengig thalassemi (eng.: transfusion dependent thalassemia – TDT), men foreløpig brukes begge deler.
Symptomer/funn ved ubehandlet thalassemia major:
- Blekhet
- Ernæringsproblemer
- “Failure to thrive”
- Infeksjonstendens
- Mikrocytær anemi
- Splenomegali
- Hos litt større barn: skjelettdeformiteter forårsaket av benmargsekspansjon, ”facies thalassaemica”
- Foci med ekstramedullær erythropoiese, spesielt langs columna
- Etter hvert utvikling av hjertesvikt (pga. økende anemi og plasmaekspansjon)
- Vanligvis død innen 5 år
Transfusjonsbehandling ved thalassemia major
β-thalassemia major behandles med transfusjoner som skal startes så snart diagnosen er stilt, og det er klart at det dreier seg om en transfusjonsavhengig fenotype. Målet med et regelmessig transfusjonsprogram er først og fremst å sikre normal vekst og utvikling, dernest å supprimere den ineffektive erythropoiesen slik at sekundære skjelettforandringer kan unngås. Før oppstart av et transfusjonsprogram anbefales det å observere pasienten over en periode på minst 3 måneder for å fastslå om det virkelig foreligger transfusjonsavhengighet. Oppstart av et transfusjonsprogram har så store konsekvenser at det er av avgjørende betydning at barnet er observert lenge nok til at man er sikker på at transfusjoner er nødvendig for å opprettholde normal vekst og trivsel. Kliniske symptomer («failure to thrive», spiseproblemer) eller sekundære forandringer (splenomegali, skjelettforandringer) forsterker indikasjonen for transfusjonsbehandling. Pga. utvikling av sekundærforandringer vil det hos noen barn bli indikasjon for å starte transfusjoner senere, selv om de i utgangspunktet har en intermediafenotype.
Det er publisert retningslinjer (guidelines) fra ulike land for behandling av pasienter med thalassemi. De er relativt sammenfallende, dog med detaljforskjeller (Musallam mfl. 2013). Felles for dem er at de alle understreker at oppstart av et transfusjonsprogram må basere seg på sikker thalssemidiagnose, kronisk anemi ledsaget av anemisymptomer, og fravær av andre forklaringer på anemien. Hemoglobingrensen for å starte transfusjonsbehandlingen angis til enten tre målinger i «steady state» ≤ 7 g/dL (Cunningham mfl. i Nathan & Oski 2009, Musallam mfl.2013) eller tre målinger ≤ 6 g/dL (Olivieri og Weatherall i Arceci, Hann, & Smith 2006, Vichinsky mfl. 2012). Hemoglobinverdien må alltid ses i sammenheng med det kliniske bildet for øvrig. I en del tilfeller kan det være riktig å gi en enkelt transfusjon først, og deretter reevaluere etter en observasjonsperiode. Når et transfusjonsprogram først er startet, anbefales det at pretransfusjons-Hb bør være mellom 9 og 10 g/dL. Optimal pretransfusjons-Hb har vært omdiskutert, men studier viser at en pretransfusjons-Hb mellom 9 og 10 g/dL er i stand til å supprimere den ineffektive erythropoiesen tilstrekkelig til å unngå sekundære skjelettforandringer (Cazzola mfl. 1995). Ved høyere pretransfusjons-Hb vil total tilført blodmengde og dermed jernbelastningen bli større (Cazzola mfl. 1997), og noen anbefaler derfor at pretransfusjons-Hb ikke bør overstige 9.5 g/dL (Olivieri 1997).
Før første transfusjon må det utføres utvidet blodtyping for å minimalisere risikoen for immunisering. Det må også gjøres en nøye serologisk utredning med henblikk på virusantistoffer (hepatitt B og C, HIV). Det må bare benyttes filtrerte blodprodukter, og om mulig er det ønskelig å gi blod fra færrest mulig blodgivere.
Transfusjonsprogram ved thalassemi:
|
På grunn av behovet for forutsigbarhet hos pasienter som transfunderes regelmessig, vil pretransfusjons-Hb kunne variere noe fra gang til gang. Dette kan kompenseres ved å variere transfusjonsvolumet og/eller endre transfusjonshyppigheten. En praktisk tilnærming til transfundert volum kan være:
- Hb < 9.0: gi 20 mL/kg
- Hb 9.0–10.0: gi 15 mL/kg
- Hb > 10.0: gi 10 mL/kg (eller vente noen dager hvis det er praktisk mulig)
Risikoer ved transfusjonsbehandling
Disse er først og fremst risiko for transfusjonsreaksjoner, transfusjonsoverførte infeksjoner og jernopphopning. Risikoen for transfusjonsreaksjoner reduseres ved utvidet blodtyping før første transfusjon, men er likevel signifikant. Risikoen er økt ved oppstart av transfusjoner etter de første 3–5 leveårene. Infeksjoner som følge av transfusjoner er sjelden, men risikoen kan aldri elimineres fullstendig.
Komplikasjoner til jernopphopning:
- Hjerte: arytmier, hjertesvikt
- Lever: fibrose → cirrhose
- Endokrine organer: hypogonadotrop hypogonadisme (forsinket eller manglende spontan pubertetsutvikling), hypothyreose, veksthormonmangel, diabetes mellitus
Behandling av jernopphopning omtales nedenfor.
Splenektomi
En del pasienter med thalassemi utvikler hypersplenisme, selv om dette i stor utstrekning kan avverges ved et godt gjennomført transfusjonsregime. Hypersplenisme øker transfusjonsbehovet, og det må derfor føres regnskap over tilført blodmengde og vektutvikling slik at man kan regne ut årlig transfusjonsbehov i forhold til vekt. Hvis årlig transfusjonsbehov overskrider ca. 250–300 ml SAG-erythrocytter/kg (gjennomsnittsvekt i løpet av året), skyldes det sannsynligvis enten utvikling av erythrocyttantistoffer eller hypersplenisme. Vurder samtidig om miltstørrelsen har økt, og ev. leukopeni/trombocytopeni. Splenektomi bør som alltid ellers unngås de første leveårene (før ca. 5-årsalder).
Indikasjonene for splenektomi er blitt betydelig innskjerpet de siste årene, da det er blitt klart at risikoen for pulmonal hypertensjon og trombose øker sterkt etter splenektomi. Før eventuell splenektomi må pasienten vaksineres mot pneumokokker og meningokokker og deretter få forebyggende penicillinbehandling etter vanlige retningslinjer.
Jernkelerende behandling
Siden kroppen ikke har noen utskillelsesmekanisme for jern, vil regelmessige transfusjonermedføre oppbygging av et jernoverskudd i kroppen. Overskuddsjern vil etter hvert avleires i parenchymatøse organer og skade disse. Mest alvorlig er avleiring i myokard som forårsaker kardiomyopati som på lang sikt kan gi arytmier og hjertesvikt. Hjertesvikt som følge av jernopphopning er den viktigste dødsårsaken hos veltransfunderte thalassemipasienter. Andre komplikasjoner til jernoverskudd er leverfibrose/-cirrhose og endokrine forstyrrelser (gonadotropinmangel med forsinket pubertet, ev. fravær av spontan pubertetsutvikling, hypothyreose, diabetes mellitus, veksthormonmangel).
Jernoverskudd må fjernes med jernkelerende behandling. Målet med jernkelerende behandling er å redusere de toksiske effektene av jern på organene, men samtidig holde bivirkningene av behandlingen så beskjedne som mulig. Den vanlige anbefalingen tidligere har vært å redusere kroppens jerninnhold til et nivå omtrent som det man finner hos personer som er heterozygote for mutasjon for hemokromatose (Olivieri 1997).
I de senere år, etter at man har fått flere jernkelerende medikamenter å spille på, anbefaler mange å tilstrebe en mer intensiv kelering med mål å få serum-ferritin ned mot normalområdet, eventuelt ved bruk av kombinasjonsbehandling (Farmaki mfl. 2009, Berdoukas mfl. 2011, Kwiatkowski 2011). Farmaki-studien beskriver reversering av hjertesvikt og endokrine forstyrrelser ved langvarig intensiv kelering som har brakt kroppens totale jerninnhold ned i normalområdet.
Monitorering av jernoverskudd
Kroppens jernlagre monitoreres rutinemessig ved måling av serum-ferritin, men dette er en dårlig parameter fordi ferritin også påvirkes av mange andre faktorer (akuttfasereaksjoner, leversykdom). Enkeltmålinger av ferritin har derfor liten verdi, men tendensen over tid er nyttig å følge. Det er dokumentert en klar assosiasjon mellom serum-ferritinverdier og langtidsoverlevelse, med økt risiko for hjertesykdom og tidlig død hos pasienter som har ferritinverdier > 2500 μg/L over lengre tid (Olivieri 1994, Borgna-Pignatti 2004). Gullstandarden for måling av kroppens jerninnhold har vært leverbiopsi med kvantitering av leverens jernkonsentrasjon, men dette har ikke vært vanlig å gjøre i Norge annet enn før stamcelletransplantasjon, og det er også internasjonalt i stor utstrekning forlatt til fordel for MRundersøkelser. Det er vist at det er dårlig samsvar mellom leverens og myokards jerninnhold.
Det er mulig å kvantitere jerninnholdet i lever og myokard ved forskjellige MR-teknikker, der MR T2* (Anderson mfl. 2001, Hershko mfl. 2004) er særlig velegnet til å fremstille jerninnholdet i myokard. MR T2* er nå tilgjengelig ved alle landets regionsykehus. Det anbefales at denne undersøkelsen inngår i monitoreringen fra 8–10-årsalder hos barn som transfunderes regelmessig, og hos barn med økt risiko for jernopphopning pga. ineffektiv erythropoiese (β-thalassemia intermedia, HbH-sykdom, enkelte andre tilstander). Jerninnholdet i lever monitoreres bedre ved R2 MRI (Ferriscan) som foreløpig ikke utføres i Norge.
Når bør behandlingen starte?
Jernkelerende behandling bør starte før det er kommet kliniske tegn på jerntoksisitet. Det er i Norge tre godkjente medikamenter til behandling av transfusjonsbetinget jernoverskudd: deferoksamin (Desferal®, DFO), deferipron (Ferriprox®, DFP) og deferasirox (Exjade®, DFX). Av disse er bare deferoksamin godkjent som førstehåndsbehandling hos barn under 6 år, så det bør i de fleste tilfellene være første valg hos de minste barna. I praksis har det de siste årene vist seg at mange velger å starte behandlingen med deferasirox, siden den er lettere å gjennomføre.
Det har vært anbefalt å ikke starte behandling med DFO før etter fylte 3 år fordi vekstforstyrrelser som komplikasjon til DFO er hyppigst hos små barn (De Virgilis et al 1988, Porter 2002). Det er imidlertid ofte konflikt mellom antall transfusjoner og ferritinverdi på den ene siden og alder på den annen, slik at man kan være nødt til å finne et kompromiss. Ved kvantitering av leverens jerninnhold er det påvist jernopphopning i leveren hos transfunderte barn allerede etter 10–12 transfusjoner, og på denne bakgrunn anbefaler de fleste at jernkelerende behandling startes etter 10–12 transfusjoner eller ca. 1 års regelmessig transfusjonsbehandling, selv om barnet ikke har fylt 3 år (Olivieri 1997). Vekstforstyrrelser ved behandling med DFO er doserelatert, slik at hvis det er indikasjon for å starte behandling før 3-årsalder, er det spesielt viktig å holde dosene i nedre anbefalte område. En mulighet er å starte med intravenøs DFO i tilslutning til transfusjonene før man introduserer de subcutane infusjonene. DFO kan gis samtidig med pågående transfusjon, men bør gis over lengre infusjonstid enn blodet, gjerne som infusjon over 24 timer.
1. Behandling med deferoksamin (Desferal®)
Deferoksamin (DFO, Desferal®) har vært brukt i behandlingen av jernoverskudd i bortimot 50 år og var lenge det eneste godkjente medikamentet til jernkelerende behandling. Behandling med DFO har medført en markert bedring i overlevelsen av pasienter med β-thalassemia major (Borgna-Pignatti mfl. 2004). DFO har svært kort halveringstid (20 minutter), absorberes ikke etter peroral administrasjon og bør derfor gis som infusjon over lang tid (i praksis som regel 8–12 timer). Det gir jernutskillelse i urin og avføring.
Gjennomføring av deferoksaminbehandling:
|
Toksisitet av deferoksamin
- Lokal smerte og inflammasjon på infusjonsstedet ved sc infusjon. Tiltak: Øke fortynningen av DFO, ev. tilsette 5–10 mg hydrokortison til infusjonsløsningen.
- Vekstforstyrrelser, spesielt hos små barn og ved høye enkeltdoser. Reduksjon først og fremst i sittehøyde.
- Høyfrekvent hørselstap, tinnitus.
- Øyeforandringer: retinaforandringer med nedsatt mørkeadaptasjon, sentralt synsfeltutfall, patologisk ERG. Katarakt. Synsforstyrrelser indiserer midlertidig seponering av DFO.
- Yersinia-infeksjoner. Mistanke om dette indiserer midlertidig seponering av DFO.
Generelt gjelder at toksisiteten av DFO er mest uttalt ved høye doser og ved små jernlagre (Hoffbrand mfl. 2012), og man bør derfor redusere dosen hvis ferritin synker < 1000 μg/L, og ta en behandlingspause hvis ferritin synker < 500 μg/L. Samtidig er det viktig å være klar over at det er beskrevet både toksiske nivåer av jern i myokard og endokrin dysfunksjon hos pasienter som har fått behandling med DFO fra ung alder, og som har relativt lave ferritinverdier, noe som understreker betydningen av å supplere med andre målemetoder for å vurdere kroppens jernoverskudd.
Det har vært anbefalt å bruke en toksisitetsindeks for dosering av DFO definert som gjennomsnittlig daglig DFO-dose (mg/kg) dividert med S-ferritin (μg/L). Denne bør ikke overstige 0.025 (tilsvarer en gjennomsnittsdose på 25 mg/kg DFO daglig ved S-ferritin på 1000 μg/L). Dette er lite omtalt i nyere litteratur, men er fremdeles nevnt i 3. utgave av UK guidelines fra 2016.
Hovedproblemet med behandlingen med DFO er dårlig etterlevelse pga. ubehaget forbundet med behandlingen. Derfor er det fremdeles en høy mortalitet på grunn av hjertesvikt forårsaket av transfusjonsbetinget jernoverskudd hos pasienter som ikke har klart å gjennomføre den jernkelerende behandlingen optimalt. Ved manifest hjertesvikt på grunn av jernoverskudd er intensivert behandling med DFO dokumentert å kunne gi rask jernutskillelse, reversering av symptomene og på lang sikt også normalisering av hjertefunksjonen hos de fleste pasientene (Cohen mfl. 1989, Anderson mfl. 2004). I slike situasjoner vil man i dag heller anbefale kombinasjonsbehandling med et av de andre medikamentene, se under punkt 4.
2. Behandling med deferipron (Ferriprox®)
Deferipron (DFP) er et peroralt jernkelerende medikament som er registrert som annenhånds middel til pasienter med thalassemia major der behandling med deferoksamin er «kontraindisert eller utilstrekkelig» (preparatomtale). Preparatet finnes som tabletter og mikstur. Det har middels lang halveringstid (2–3 timer), doseres peroralt, 25 mg/kg 3 ganger daglig før måltid (kan økes til 33 mg/kg x 3), gir jernutskillelse vesentlig i urinen, men gir dårligere reduksjon i leverens jerninnhold enn deferoksamin. En rekke studier viser at deferipron er spesielt effektivt til å fjerne jern fra myokard (Anderson et al 2002, Borgna-Pignatti et al 2006, Pennell et al 2006). Dette forklares med at DFP er et lite molekyl som er lipofilt, og derfor lett passerer cellemembranen og binder jern intracellulært.
Den mest alvorlige bivirkningen av deferipron er agranulocytose (nøytrofile < 0.5 x 109/L) og nøytropeni (nøytrofile 0.5–1.5 x 109/L) som er beskrevet i hhv. 0.5–1.8 og 2.1–5.4 tilfeller per 100 pasientår (Ceci 2002) eller hhv. < 1 og 8 % av pasientene, noe sjeldnere hos splenektomerte (Cohen 2006). Agranulocytose er en reversibel bivirkning, men indiserer seponering av medikamentet. Bruk av DFP krever derfor hyppig blodprøvekontroll, ifølge fabrikanten ukentlig og ved feber, men kan nok ved god toleranse justeres til hver annen uke etter det første året (Hoffbrand mfl. 2012). Etter at medikamentet ble godkjent og dermed er kommet i mer utstrakt bruk, har det vært rapportert flere dødsfall som følge av agranulocytose. Flere av dødsfallene har vært hos pasienter som har fått DFP på ikke-godkjent indikasjon (ulike former for benmargssvikt), og hos alle manglet data på ukentlig telling av hvite blodlegemer.
Toksisitet av deferiprone:
- Agranulocytose eller nøytropeni (sjelden)
- Arthralgier (relativt hyppig, inntil 20 %)
- Gastrointestinalt ubehag (som regel forbigående uten dosereduksjon)
- Transaminaseøkning
- Fall i S-sink
3. Behandling med deferasirox (Exjade®)
Deferasirox (DFX) ble registrert høsten 2006 som jernkelerende medikament til pasienter med transfusjonsbetinget jernoverskudd, til å begynne med i form av dispergerbare tabletter, senere erstattet av filmdrasjerte tabletter. (De to administrasjonsformene har ulik dosering, så det er viktig når man leser internasjonal litteratur, å være klar over hvilket preparat som omtales.) DFX har lang halveringstid (8–16 timer) og kan derfor doseres én gang daglig, og det har den store fordelen at det gir kelerende effekt 24 timer i døgnet. Jernutskillelsen ved bruk av DFX ervesentlig gjennom avføringen. Vanlig startdose er 14 mg/kg daglig (tilsvarer 20 mg/kg av dispergerbare tabletter), maksimal dose angis til 28 mg/kg daglig (tilsvarer 40 mg/kg av dispergerbare tabletter). Preparatet er godkjent ved transfusjonsbetinget jernoverskudd for barn over 2 år, som førstehåndspreparat over 6 år som beskrevet ovenfor. Dersom serum-ferritin faller < 1000 μg/L, bør DFX-dosen reduseres.
Det var lenge usikkerhet rundt effekten av deferasirox på jerninnholdet i myokard, men det er etter hvert publisert flere studier som dokumenterer at også DFX er effektivt til å fjerne jern fra hjertet (Pennell mfl. 2012, Cassinerio mfl. 2012).
Toksisitet av deferasirox:
- Kreatininstigning (uten tegn på progredierende nyresvikt)
- Gastrointestinale plager – ofte forbigående
- Transaminaseøkning
- Utslett – forbigående
- Renalt Fanconi-syndrom – proximal tubulær dysfunksjon (Rheault mfl. 2011, Chuang mfl. 2015)
Når man starter behandling med DFX, vil forsiktig opptrapping av dosen (fra 7 mg/kg daglig) redusere risikoen for gastrointestinale bivirkninger og utslett.
| Det har alltid vært kontroversielt hvilket peroralt preparat som er å foretrekke, dels begrunnet i ulik vurdering av effekt, dels i toksistetsprofil. Deferipron ble godkjent i Europa allerede i 1999, etter langvarig bruk som generisk preparat (L1) i India (Agarwal 1992) og etter studier ved Royal Free Hospital i London fra 1987 (Kontoghiorghes mfl. 1987, Hider mfl. 2018). Preparatet ble godkjent i USA i 2011 og i Canada i 2015. Etter hvert har de fleste funnet en balanse i hvordan man kan utnytte de ulike preparatenes egenskaper til å skreddersy keleringsbehandlingen til hver enkelt pasient. Kontroversen har nylig blusset opp igjen etter nye studier av både effekt og toksistet (Kontoghiorghe mfl. 2016, Olivieri mfl. 2019, Schafer 2020, Badeli mfl. 2019, Chuang mfl. 2015). Her reises også spørsmålet om forskernes (u)avhengighet av sponsorene i publiseringen av resultatene (Schafer 2020). Dette understreker igjen behovet for nitid monitorering av alle pasienter som behandles med jernkelerende medikamenter, slik at man kan optimalisere behandlingen best mulig for hver enkelt pasient. |
4. Kombinasjonsbehandling
Det har i de senere årene blitt publisert en rekke studier som tar for seg kombinasjonsbehandling med de ulike jernkelerende medikamentene (oppsummert i Cappellini, TDT guidelines 2014). Best undersøkt er kombinasjonen deferoksamin/deferipron som ser ut til å være det mest effektive hvis målet er å redusere jernopphopningen raskt (Tanner mfl. 2007, 2008), f. eks. ved hjertesvikt, som forberedelse til stamcelletransplantasjon eller før planlagt svangerskap. Det er beskrevet en «shuttle effect» som innebærer at det lille DFP-molekylet passerer cellemembranen, binder seg til jern intracellulært og tar det med seg ut i sirkulasjonen der det kan overføres til DFO og skilles ut (Evans mfl. 2010, Hoffbrand mfl. 2012). Man tenker seg at DFP deretter kan «gjenbrukes». Kombinasjonen er mest effektiv om DFO gis som kontinuerlig iv infusjon 24 timer i døgnet.
Men også kombinasjoner av DFO/DFX (Lal mfl. 2013, Aydinok mfl. 2015) og DFP/DFX (Elalfy mfl. 2015, Totadri mfl. 2015, Karami mfl. 2017) er nå publisert og funnet effektive og med god sikkerhetsprofil.
Kombinasjonsbehandling har også den fordelen at man kan bruke lavere doser av hvert medikament og derved redusere bivirkningene (Hoffband mfl. 2012)
Oppfølging av pasienter under transfusjons-/jernkelerende behandling
- Ved hver transfusjon
- Full klinisk undersøkelse
- Hematologisk status med diff., pretransfusjonsprøve. Andre prøver på indikasjon
- Samtale med sykepleier om gjennomføringen av den jernkelerende behandlingen
- Hver 3. måned
- Høyde- og vektregistrering, inntegning i percentilskjema
- Faste blodprøver + Na, K, Ca, Mg, fosfat, sink, transaminaser, ALP, LD, bilirubin, kreatinin, syre-base
- S-ferritin, beregning av toksisitetsindeks for DFO, evt. dosejustering
- Urin-stix (glucose), evt. fosfat, kreatinin, protein-kreatinin-ratio, calcium, aminosyrer (ved DFX-behandling)
- Årlig
- Blodprøver: faste + hver 3. måned + vitamin D-status, PTH, screening for blodbårne infeksjoner på indikasjon (hvis transfundert i utlandet)
- Stå- og sittehøyde
- Transfusjonsregnskap, utregning av total transfundert blodmengde pr. kg (gjennomsnittsvekt gjennom året)
- UL abdomen med henblikk på miltstørrelse
- Rtg. thorax med henblikk på hjertestørrelse og eventuell forekomst av ekstramedullære hematopoietiske foci
- Kardiologisk vurdering: EKG, ekko-Doppler med henblikk på venstre ventrikkels ejeksjonsfraksjon
- Glukosebelastning med henblikk på prediabetes
- Audiometri (DFO-toksisitet)
- Øyeundersøkelse med synsvurdering og øyebunnsundersøkelse (DFO-toksisitet)
- Fra ca. 10 års alder og senere årlig
- Pubertetsstadium: Tanner
- Hormonstatus: FSH, LH, østradiol, testosteron, T3, T4, TSH. Veksthormon, IGF-1 og rtg. skjelettalder på klinisk indikasjon
- MR T2* av lever og myokard (fra 8–10-årsalder)
- Hvis normal (> 20 ms): gjentas etter 2–3 år
- Hvis patologisk: intensiver jernkeleringen, kontroll etter 3-6-12 måneder
- Sentral oppfølging
Det anbefales årlig kontroll ved eller i samarbeid med senter med særlig kompetanse innen hemoglobinsykdommer
Stamcelletransplantasjon
Thalassemi kan helbredes med hematopoietisk stamcelletransplantasjon (SCT). Det er oppnådd gode resultater med SCT med forlikelig søskengiver, med mellom 80 og 90 % thalassemifri overlevelse avhengig av risikoklasse2 (Gaziev og Lucarelli 2005). Etter hvert som man har modifisert forbehandlingen i retning av mindre toksiske kondisjoneringsregimer, er det også oppnådd sammenlignbare resultater med ubeslektet donor (Bernardo mfl. 2012). I Norge har thalassemia major vært godkjent som transplantasjonsindikasjon for barn med bruk av forlikelig familiegiver i mange år, men det er nå også åpnet for transplantasjon med ubeslektet giver etter individuell vurdering.
Etter en vellykket SCT må behandlingen for å redusere kroppens jernoverskudd fortsette. Dette kan skje med fortsatt DFO eller deferasirox (deferipron anbefales ikke etter SCT pga. uavklart risiko for nøytropeni/agranulocytose i den situasjonen), men oftest anbefales venesectio som den beste behandlingen for å redusere jernoverskuddet etter transplantasjon. Venesectio kan starte ca. 1 år etter transplantasjonen forutsatt normal erythropoietisk aktivitet og fortsette til ferritin er under ca. 300-400 (hos voksne anbefales å fortsette til ferritin er < 100) (Angelucci, UpToDate 2016).
Genterapi/genredigering
Det har i mange år vært gjort omfattende forskning med sikte på å helbrede pasienter med ulike genetiske sykdommer, deriblant β-thalassemia major, ved hjelp av genterapi (High og Roncarolo 2019). Det har nå kommet data fra flere kliniske studier (Cao mfl. 2011, de Dreuzy mfl. 2016,
2 Risikofaktorer: leverforstørrelse > 2 cm nedenfor arcus, leverfibrose påvist ved leverbiopsi, dårlig gjennomført jernkelerende behandling.
Risikoklasse 1: ingen risikofaktorer
Risikoklasse 2: 1-2 risikofaktorer
Risikoklasse 3: 3 risikofaktorer
Cappellini mfl. 2018, Thompson mfl. 2018), og resultatene så langt ser lovende ut, men behandlingen er fremdeles i tidlig fase.
Det er to hovedprinsipper i behandlingen (Cappellini mfl. 2018):
- Genterapi: Man høster og isolerer autologe stamceller/hematopoietiske progenitorceller og introdusere deretter, ved hjelp av en lentivirusvektor, normale eksogene β- eller γglobingener i cellene in vitro. Pasienten gjennomgår så myeloablativ kondisjonering før de endrede stamcellene tilbakeføres. Gevinsten av behandlingen må veies opp mot toksisiteten av myeloablativ kondisjonering.
Det er nylig EMA3-godkjent et kommersielt produkt (Zynteglo® [autologous CD34+ cells encoding βA-T87Q-globin gene]) som ved hjelp av en lentivirusvektor kan overføre et fungerende β-globingen til pasientens egne stamceller, som så kan tilbakeføres til pasienten etter myeloablativ kjemoterapi. Produktet er ikke beregnet for homozygote (β0β0) pasienter fordi resultatene ikke har vært gode nok i denne gruppen. Det avventes data fra post-marketing-studier. - Genredigering: Man tilstreber å korrigere den spesifikke genfeilen i β-globingenet ved hjelp av designer-nukleaser. Alternativt kan man stimulere økt HbF-produksjon, enten ved å stimulere γ-globin-transkripsjonsfaktorer, eller oppheve hemmingen av HbFproduksjonen, slik at denne vedvarer permanent.
Alternativ medikamentell behandling
Det arbeides med å fremstille medikamenter som kan redusere den ineffektive erythropoiesen (Cappellini mfl. 2018, Porter 2018, El-Beshlawi og El-Ghamrawy 2019).
- Hydroksyurea (HU – hydroksykarbamid) er et gammelt medikament som er kjent for å gi økt produksjon av HbF, en egenskap som utnyttes i behandlingen av sigdcellesykdom. Ved thalassemi er effekten uforutsigbar, men en del pasienter med thalassemia intermedia (først og fremst HbEβ0 ) kan ha til dels god nytte av det, eventuelt i kombinasjon med erythropoietin (EPO), se nedenfor.
- JAK2-hemmeren ruxolitinib er et medikament som brukes i behandlingen av polycytemia vera, og som har vært testet ved transfusjonsavhengig thalassemi. Det har ikke vært vist bedring av pretransfusjons-Hb eller reduksjon i transfusjonsbehovet hos pasienter med TDT, så det planlegges ikke flere studier med dette medikamentet.
- Sotatercept og luspatercept er fusjonsproteiner for hhv. aktivin-reseptor IIA og IIB. Begge reduserer presipiteringen av løse α-globinkjeder i sene erythrocyttforstadier, og reduserer derved den intramedullære hemolysen/ineffektive erythropoiesen. Begge har vist bedret Hb ved non-transfusjonsavhengig thalassemi og reduksjon i transfusjonbehovet ved transfusjonsavhengig thalassemi i preliminære studier.
- Hepcidin-analoger er under utprøving. De virker ved å redusere tilbudet av jern til normoblastene.
β-thalassemia intermedia (TI)
I utgangspunktet er pasienter med β-thalassemia intermedia ikke avhengige av regelmessige transfusjoner for å opprettholde normal vekst og utvikling (non-transfusjonsavhengig thalassemi – NTDT). Disse pasientene kan utransfundert opprettholde Hb-verdier > ca. 7 g/dL i hvert fall de 3 EMA- European Medicines Agency første leveårene, selv om sporadiske transfusjoner kan bli nødvendig. For differensieringen mellom thalassemia major og intermedia, se ovenfor.
Pasienter med β-thalassemia intermedia har varierende grad av ineffektiv erythropoiese og har derfor tendens til benmargsekspansjon, skjelettdeformiteter og iblant ekstramedullær erythropoiese som vil kunne indisere oppstart av et transfusjonsprogram litt senere enn det som er nødvendig ved thalassemia major. Pasientene har som regel økt jernabsorpsjon fra tarmen, og selv uten regelmessige transfusjoner kan den økte jernabsorpsjonen føre til klinisk betydningsfull jernopphopning.
Pasienter med β-thalassemia intermedia har økt risiko for trombose og for utvikling av pulmonal hypertensjon (Karimi mfl. 2011). Det er påvist en generell økt hyperkoagulabilitet som er mer uttalt hos utransfunderte TI-pasienter enn hos transfunderte TM-pasienter (Cappelini mfl. 2010, 2012). Den økte risikoen tilskrives en kombinasjon av flere faktorer: ødelagte erythrocytter/ erythrocyttfragmenter og kjerneholdige røde blodceller i perifert blod, høyt antall trombocytter, endotelaktivering, pulmonal hypertensjon. Risikoen øker betydelig etter splenektomi, og det er derfor også hos TI-pasienter grunn til å være tilbakeholden med splenektomi. Det er ikke data på bruk av antikoagulasjonsbehandling eller anti-trombocytt-medikamenter, men det er beskrevet en mulig beskyttende effekt av acetylsalicylsyre som sekundærprofylakse hos pasienter som har gjennomgått en trombotisk episode (Taher mfl. 2006).
HbEβ0-thalassemi har et svært varierende klinisk forløp, men arter seg oftest som thalassemia intermedia. Denne thalassemivarianten er kanskje den mest utbredte formen for thalassemi på verdensbasis, spesielt i Sydøst-Asia (Weatherall 2001).
Behandling
Pasientene må følges nøye med kliniske undersøkelser og monitorering av hematologisk status og jernhusholdning. Unngå unødvendig jernbehandling eller kosttilskudd som inneholder jern. På grunn av ineffektiv erythropoiese er det behov for ekstra folsyretilskudd, minst 1 mg daglig. Tradisjonelt har det vært anbefalt jernfattig kost, dette er av større betydning ved thalassemia intermedia enn major fordi transfusjonene alltid vil utgjøre en mye større tilført jernmengde enn kosten. I noen tilfeller kan jernkelerende behandling bli nødvendig, også hos pasienter som ikke har fått regelmessige transfusjoner.
Sekundære skjelettforandringer, tegn på ekstramedullær erythropoiese eller økende anemi kan være indikasjon for å starte transfusjoner litt opp i barnealder. Et transfusjonsprogram startet på dette grunnlag behøver ikke alltid være livslangt, og man kan forsøksvis trappe ned transfusjonene med sikte på transfusjonsfrihet etter at veksten er avsluttet. Økende anemi kan også være uttrykk for hypersplenisme, og splenektomi kan i så fall overveies. På grunn av den sterke økningen i risiko både for trombose og for utvikling av pulmonal hypertensjon etter splenektomi, en risiko som er større hos pasienter med TI enn TM, må indikasjonen for splenektomi være sterk og veloverveid.
Medikamentell behandling for å øke produksjonen av HbF kan i noen tilfeller ha effekt, best dokumentert ved HbEβ0-thalassemi (Singer mfl. 2005). Effekten er uforutsigelig og overveiende dårligere enn ved sigdcelleanemi (Steinberg 2001, Fathallah mfl. 2005, Singer mfl. 2005, de Dreuzy mfl. 2016). Det mest aktuelle medikamentet har så langt vært hydroksyurea (HU – hydroksykarbamid, i dosering 15–20 mg/kg daglig). I noen studier er det funnet at HU reduserer risikoen for trombotiske og endokrine komplikasjoner og ekstramedlullær erythropoiese, uavhengig av effekten på Hb (Musallam mfl. 2013). Effekten på hyperkoagulabilitet er mest uttalt hos splenektomerte pasienter.
Effekten av hydroksyurea på erythropoiesen øker ved samtidig behandling med erythropoietin(EPO) (Elalfy mfl. 2013). Ved behandling med EPO kan det bli behov for jerntilførsel for å sikre tilstrekkelig tilgjengelig jern til erythropoiesen, selv om pasientene i utgangspunktet kan ha jernoverskudd. Butyrat kan også i noen tilfeller øke produksjonen av HbF.
Oppfølging
Siden pasienter med TI opplever mange av de samme komplikasjonene som TM-pasienter, må de også følges relativt tett opp med utgangspunkt i oppfølgingsprogrammet skissert for TMpasienter. De må følges med henblikk på vekst, jernhusholdning, mulig forverring av anemien, endokrine forstyrrelser etc. Det må tas stilling til utvikling av eventuelt senere transfusjonsbehov, eventuelt gjøre et behandlingsforsøk med hydroksyurea. Indikasjonen for splenektomi må også vurderes, men splenektomi bør helst unngås, se ovenfor.
β-thalassemia minor
β-thalassemia minor er å betrakte som en arvebærertilstand. Differensialdiagnose: jernmangel. Tilstanden krever ingen behandling, men det er viktig at affiserte personer får genetisk veiledning.
Prenataldiagnostikk/screening
Prenataldiagnostikk er mulig, ved chorionbiopsi og DNA-undersøkelser ved kjent gendefekt, ellers ved føtal blodprøve. Screening med henblikk på thalassemi er mest aktuelt i form av prekonsepsjonsscreening eller screening av gravide kvinner, siden nyfødtscreening ved denne tilstanden ikke vil ha umiddelbar terapeutisk konsekvens (i motsetning til ved sigdcelleanemi). I land med høy prevalens av thalassemi har populasjonsbaserte antenatale screeningprogrammer gitt dramatisk reduksjon i fødselsraten av affiserte barn (Davies et al 2000, Leung mfl. 2005).
Screening for thalassemi innebærer undersøkelse av MCV og/eller MCH, og ved lave verdier (MCV < 80, MCH < 27) går man videre med undersøkelse av hemoglobinmønsteret og eventuelt DNA-undersøkelser. Ved jernmangel må undersøkelsen eventuelt gjentas etter behandling av jernmangelen, da jernmangel hemmer økningen av HbA2 som man vanligvis ser ved βthalassemia minor. HbA2 > 3.5 % oppfattes som diagnostisk for β-thalassemia minor. MCV stiger ved lagring av blodprøvene, og av den grunn er nok MCH bedre egnet til screening.
Overføring til voksenavdeling
Overføring til behandling og oppfølging i voksenavdeling bør skje gradvis og må forberedes i god tid. Siden pasientantallet foreløpig er relativt lite i Norge, har vi nok ikke klart å innarbeide gode overføringsrutiner. Mange voksenavdelinger har fremdeles liten erfaring med denne pasientgruppen, selv om det har bedret seg de siste årene. I UK guidelines er det anbefalt å starte overføringsprosessen allerede ved 13 års alder, og det er anbefalt at det i forbindelse med overføringen utarbeides en skriftlig individuell plan i fellesskap mellom barneavdelingen, vokseavdelingen og pasienten selv.
Referanser
- Aessopos A mfl. Correlation of echocardiography parameters with cardiac magnetic resonance imaging in transfusion-dependent thalassemia major. Eur J Haematol 2006; doi: 10.1111/j.1600- 0609.2006.00770.x
- Agarwal MB mfl. Long-term assessment of efficacy and safetyof L1, an oral iron chelator, in transfusion dependent thalassaemia: Indian trial. Br J Haematol 1992; 82: 460-466
- Anderson LJ mfl. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J 2001; 22: 2171-2179
- Anderson LJ mfl. Comparison of effects of oral deferiprone and subcutaneous desferrioxamine on myocardial iron concentrations and ventricular function in beta-thalassaemia. Lancet 2002; 360: 516-520
- Anderson LJ mfl. Myocardial iron clearance during reversal of siderotic cardiomyopathy with intravenous desferrioxamine: a prospective study using T2* cardiovascular magnetic resonance. Br J Haematol 2004; 127: 348-355
- Arceci RJ, Hann IM, Smith OP. Pediatric Hematology. Blackwell Publishing, 3rd edition 2006
- Angelucci E. Long-term mangaement of the thalassemic patient after hematopoietic cell transplantation. https://www.uptodate.com/contents/long-term-management-of-the-thalassemicpatient-after hematopoietic-cell-transplantation. Aksessert 16.11.2016
- Aydinok Y mfl. Effects of deferasirox-deferoxamine on myocardial and liver iron in patients with severe transfsional iron overload. Blood 2015; 125: 3868-3877
- Badeli H mfl. Early kidney damage markers after deferasirox treatment in patients with thalassemia major: a case-control study. Oxidative Medicine and Cellular Longevity 2019; https://doi.org/10.1155/2019/5461617
- Bain BJ. Haemoglobinopathy diagnosis. Blackwell Publishing, 2nd edition 2006
- Berdoukas V, Farmaki K, Wood JC, Coates T. Iron chelation in thalassemia: time to reconsider our comfort zones. Expert Rev Hematol 2011; 4: 17-26
- Bernardo ME mfl. Allogeneic hematopoietic stem cell transplantation in thalassemia major: results of a reduced-toxicity conditioning regimen based on the use of treosulfan. Blood 2012; 120: 473-476
- Borgna-Pignatti C mfl. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 2004; 89: 1187-1193
- Borgna-Pignatti C mfl. Cardiac morbidity in deferoxamine- or deferiprone-treated patients with thalassemia major. Blood 2006; 107: 3733-3737.
- Cao A, Moi P, Galanello R. Recent advances in β-thalassemias. Pediatric Reports 2011; 3: e17: 65-78
- Cappellini MD mfl. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with β-thalassemia. Blood 2006; 107: 3455-3462
- Cappellini MD mfl. Redefining thalassemia as a hypercoagulable state. Ann NY Acad Sci 2010; 1202: 231-236
- Cappellini MD mfl. Hypercoagulability in β-thalassemia: a status quo. Exp Rev 2012; 5: 505-512
- Cappellini MD mfl. Guidelines for the management of transfusion dependent thalassaemia (TDT). 3rd edition 2014
- Cappellini MD mfl. A paradigm shift on beta-thalassaemia treatment: How will we manage this old disease with new therapies? Blood Reviews 2018; 32: 300-311
- Cassinerio E mfl. Cardiac iron removal and functional cardiac improvement by different iron chelation regimens in thalassemia major patients. Ann Hematol 2012; 91: 1443-1449
- Cazzola M mfl. Relationship between transfusion regimen and suppression of erythropoiesis in βthalassaemia major. Br J Haematol 1995; 89: 473-478
- Cazzola M mfl. A moderate transfusion regimen may reduce iron loading in β-thalassemia major without producing excessive expansion of erythropoiesis. Transfusion 1997; 37: 135-140
- Ceci A mfl. The safety and effectiveness of deferiprone in a large-scale, 3-year study in Italian patients. Br J Haematol 2002; 118: 330-336
- Christoforidis A mfl. Four-year evaluation of myocardial and liver iron assessed prospectively with serial MRI scans in young patients with β-thalassaemia major: comparison between different chelation regimens. Eur J Haematol 2006; doi:10.1111/j.0902-4441.2006.t01-1-EJH3013.x
- Chuang G-T mfl. Transfusion-dependent thalassaemic patients with renal Fanconi syndrome due to deferasirox use. Nephrology 2015; 20: 931-935
- Cohen AR mfl. Rapid removal of excess iron with daily, high-dose intravenous chelation therapy. J Pediatr 1989; 115: 151-155
- Cohen AR. New advances in iron chelation therapy. Hematology 2006: 42-47
- Davies SC mfl. Screening for sickle cell disease and thalassaemia: a systematic review with supplementary research. Health Technol Assess 2000; 4(3)
- de Dreuzy E mfl. Current and future alternative therapies for beta-thalassemia major. Biomed J 2016; 39: 24-38
- De Virgilis S mfl. Deferoxamine-induced growth retardation in patients with thalassemia major. J Pediatr 1988; 113: 661-669
- Elalfy MS mfl. Therapeutic superiority and safety of combined hydroxyurea with recombinant human erythropoietin over hydroxyurea in young β-thalassemia intermedia patients. Eur J Haematol 2013; 91: 522-533
- El-Beshlawy A og El-Ghamrawy M. Recent trends in treatment of thalassemia. Blood Cells, Molecules and Diseases 2019; 76: 53-58
- Elalfy MS mfl. Efficacy and safety of a novel combination of two oral chelators deferasirox/deferiprone over deferoxamine/deferiprone in severely iron overloaded young beta thalassemia major patients. Eur J Hematol 2015; 95: 411-420
- Evans P mfl. Mechanisms for the shuttling of plasma non-transferrin-bound iron (NTBI) onto deferoxamine by deferiprone. Transl Res. 2010; 156: 55-67
- Fathallah H mfl. Pharmacological induction of fetal hemoglobin. Why haven’t we been more successful in thalassemia? Ann NY Acad Sci 2005; 1054: 228-237
- Farmaki K, Tzoumari I, Pappa C, Chouliaras G, Berdoukas V. Normalisation of total body iron load with very intensive combined chelation reverses cardiac and endocrine complications of thalassaemia major. Br J Haematol 2009; 148: 466-475
- Galanello R mfl. A prospective randomized controlled trial on the safety and efficacy of alternating deferoxamine and deferiprone in the treatment of iron overload in patients with thalassemia. Haematologica 2006; 91: 1241-1243
- Galanello R, Agus A, Campus S, Danjou F, Giardina PJ, Grady RW. Combined iron chelation therapy. Ann NY Acad Sci 2010 Aug; 1202: 79-86
- Gaziev J, Lucarelli G. Stem cell transplantation for thalassaemia. Reproductive BioMedicine Online 2005; 10: 111-115
- Hershko C mfl. Purging iron from the heart. Br J Haematol 2004; 125: 545-551
- Hider RC, Hoffbrand AV. The role of deferiprone in iron chelation. New Engl J Med 2018; 379: 2140-2150
- High KA, Roncarolo MG. Gene therapy. New Engl J Medicine 2019; 381: 455-464
- Hoffbrand AV, Taher A, Cappellini MD. How I treat transfusional iron overload. Blood 2012; 120: 3657-3669
- Karami H mfl. Combination iron chelation therapy with deferiprone and deferasirox in ironoverloaded patients with transfusion-dependent β-thalassemia major. Clinics and Practice 2017; 7:912: 11-14
- Karimi M mfl. Risk factors for pulmonary hypertension in patients with β-thalassemia intermedia. Eur J Int Med 2011; 22: 607-610
- Kontoghiorghe CN, Kontoghiorghes GJ. Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with non-transfusion-dependent thalassemia syndromes. Drug Design, Development and Therapy 2016; 10: 465-481
- Kontoghiorghes GJ mfl. 1,2-dimethyl-3-hydroxypyrid-4-one, an orally active chelator for treatment of iron overload. Lancet 1987; 10.1016/s0140-6736(87)90545-9
- Kwiatkowski JL. Real-world use of iron chelators. Hematology 2011: 451-458
- Lal A mfl. Combined chelation therapy with deferaxirox and deferoxamine in thalassemia. Blood Cels Mol Dis 2013; 50: 99-104,
- Leung TN mfl. Thalassaemia screening in pregnancy. Curr Opin Obstet Gynecol 2005; 17: 129-134
- Lilleyman J, Hann I, Blanchette V. Pediatric Hematology, 2nd edition 1999, Churchill Livingstone
- Maggio A mfl. Improving survival with deferioprone treatment in patients with thalassemia major: A prospective multicenter randomised clinical trial under the auspices of the Italian Society for Thalassemia and Hemoglobinopathies. Blood Cells Mol Dis 2009; 42: 247-251
- Martin A, Thompson AA. Thalassemias. Pediatr Clin N Am 2013; 60: 1383-1391
- Musallam KM mfl. Clinical experience with fetal hemoglobin induction therapy in patients with β-thalassemia. Blood 2013; 121: 2199-2212
- Musallam KM mfl. Cross-talk between available guidelines for the management of patients with beta-thalassemia major. Acta Haematol 2013; 130: 64-73
- Olivieri NF mfl. Survival in medically treated patients with homozygous β-thalassemia. N Engl J Med 1994; 331: 574-578
- Olivieri NF and Brittenham GM. Iron-chelating therapy and the treatment of thalassemia. Blood 1997; 89: 739-361
- Olivieri NF mfl. Single-center retrospective study of the effectiveness and toxicity of the oral iron chelating drugs deferiprone and deferasirox. PLOS ONE 2019; https://doi.org/10.1371/journal.pone.0211942
- Orkin SH, Nathan DG, Ginsburg D, Look AT, Fisher DE, Lux SE. Nathan and Oski’s Hematology of Infancy and Childhood, 5th edition 1998 and 7th edition 2009
- Pennell DJ mfl. Randomized controlled trial of deferiprone or deferoxamine in beta-thalassemia major patients with asymptomatic myocardial siderosis. Blood 2006; 107: 3738-3744
- Pennell DJ, Carpenter JP, Roughton M, Cabantchik ZI. On improvement in ejection fraction with iron chelation in thalassemia major and the risk of future heart failure. J Cardiovasc Magn Reson 2011; 13:45 doi:10.1186/1532-429X-13-45
- Pennell DJ mfl. Deferasirox for up to 3 years leads to continued improvement of myocardial T2* in patients with beta-thalassaemia major. Haematologica 2012 [Epub ahead of print] doi:10.3324/haematol.2011.049957
- Piga A mfl. High-dose desferrioxamine as a cause of growth failue in thalassemic patients. Eur J Haematol 1988; 40: 380-381
- Porter J. Beyond transfusion therapy: new therapies in thalassemia including drugs, alternate donor transplant, and gene therapy. Hematology 2018; 361-370
- Porter JB, Davis BA. Monitoring chelation therapy to achieve optimal outcome in the treatment of thalassemia. Best Practice and Research Clin Hematol 2002; 15(2): 329-368
- Rheault MN, Bechtel H, Neglia JP, Kashtan CE. Reversible Fanconi syndrome in a pediatric patient on deferasirox. Pediatr Blood Cancer 2011; 56: 674-676
- Schafer A. Institutional conflict of interest: attempting to crack the deferiprone mystery. J Med Ethics 2020; 0:1-8 doi:10.1136/medethics-2019-105498
- Singer ST mfl. Fetal haemoglobin augmentation in E/β0 thalassaemia: clinical and haematological outcome. Br J Haematol 2005; 131: 378-388
- Singer ST mfl. Single and combination drug therapy for fetal hemoglobin augmentation in hemoglobin E-β0-thalassemia: considerations for treatment. Ann NY Acad Sci 2005; 1054: 250-256
- Standard for the clinical care of children and adults with thalassaemia in the UK. 3rd edition 2016.
- United Kingdom Thalassaemia Society (https://www.stgeorges.nhs.uk/wp-content/uploads/2020/02/UKTS-adults-and-children-with-thalassaemia-guidelines-2016.pdf)
- Steinberg M, Rodgers GP. Pharmacologic modulation of fetal hemoglobin. Rev Mol Med 2001; 80: 328-344
- Taher A mfl. Prevalence of thromboembolic events among 8860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thromb Haemost 2006; 96: 488-491
- Tanner MA mfl. A randomized, placebo-controlled, double-blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonance. Circulation 2007; 115: 1876-1884
- Tanner MA mfl. Combined chelation therapy in thalassemia major for the treatment of severe myocrdial siderosis with left ventricular dysfunction. J Cardiovasc Magn Reson 2008; 10:12, doi10:1186/1532-429X-10-12
- Thompson AA mfl. Gene therapy in patients with transfusion-dependent β-thalassemia. NewEngl J Medicine 2018; 378: 1479-1493
- Totadri S mfl. The deferiprone and deferaxirox combination is efficacious in iron overloaded patients with β-thalassemia major: a prospective, single center, open-label study. Pediatr Blood Cancer 2015; doi 10,1002/pbc.25533
- Vichinsky E mfl. Standards of care guidelines for thalassemia. Children’s Hospital & Research Center Oakland 2012
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull WHO 2001; 79: 704-712
- Wonke B mfl. Combined therapy with deferiprone and desferrioxamine. Br J Haematol 1998; 103: 361-364