









Generell veileder i pediatri
7. Øvre og nedre luftveier
7.15 Primær ciliær dyskinesi (PCD)
Sist faglig oppdatert: 01.01.2017
Suzanne Crowley
Bakgrunn
PCD er en sjelden, heterogen genetisk sykdom som rammer bevegelige cilier og fører til dårlig slimmobilisering og stagnasjon av sekret i øvre og nedre luftveier og etter hvert til utvikling av bronkiektasier hos de fleste. Ciliene kan være enten statiske, kaotiske, hyperkinetiske eller fraværende, avhengig av genmutasjon. Sykdommen kalles primær ciliær dyskinesi for å skille den fra sekundær ciliær dyskinesi som skyldes cilieskade etter for eksempelviral luftveisinfeksjon. Så langt er det funnet mutasjoner i minst 36 forskjellige gener som årsak til strukturelle ciliedefekter som forårsaker PCD. Siden PCD har en hovedsakelig recessiv arv, sjeldent X-linked, er det antatt at prevalensen er høyere hos gruppersom har høy forekomst av inngifte, men også at dener likt fordelt mellom kjønnene. Den estimerte prevalensen varierer fra 1:10 000 til 1:30 000, men PCD er underdiagnostisert. Det er ca 70 kjente PCD-barn og -voksne i Norge, mens man skulle forvente minst 180 PCD-pasienter. Fordi embryonale nodale cilia også er affisert ved PCD tilsier statistisk tilfeldighet atca 50 % av pasientene vil ha situs inversus totalis (SI). Videre har 12 % heterotaxi (situs ambiguus) som gir 200 ganger økt risiko for medfødte hjertefeil. Kombinasjonen av bronkiektasier, situs inversus og manglende frontal sinus kalles Kartagener syndrom. Til tross for at nesten alle PCD-pasienter har luftveissymptomer fra første levedag eller tidlig i nyfødt perioden, diagnostiseresPCD sent i Europa; gjennomsnittlig alder ved diagnose er 5,8 år og 3,5 år hos barn med situs inversus. Det finnes sykdommer som rammer primære (ikke-bevegelige) cilier også, for eksempel OFD1 syndrom og Bardet-Biedl syndrom. Pasienter med Bardet-Biedl syndrome har økt forekomst av luftveissymptomer, men ikke motile cilier dysfunksjon. Pasienter med mutasjoner i OFD1 genet kan ha en rekke funn inkludert polydactyly, mental retardasjon, multicystisk nyresykdom og retinitis pigmentosa i tillegg til økt forekomst av luftveissymptomer. Pasienter med primære ciliopatier (i motsetning til PCD) pleier ikke å utvikle bronkiektasier.
Symptomer og funn
Minst 75 % av nyfødte med PCD har symptomer i løpet av første levedøgn, blant annet respirasjonsbesvær med behov for supplerende oksygen eller CPAP i dager/uker, pneumoni, atelektase og nesetetthet. I nyfødtperioden, vil kombinasjonen av atelektase, SI og behov for supplerende oksygen >24 timer ha 87 % sensitivitet og 96 % spesifisitet for en sikker PCD-diagnose. Nesten alle med PCD har en kronisk, daglig våt hoste fra tidlig i det første leveåret, som ikke er sesongavhengig og som vedvarer tross antibiotikabehandling. Gjentatte nedre luftveisinfeksjoner eller pneumonier er vanlige. Minst 70–80 % av PCD-pasientene har kronisk rhinosinusitt med nesetetthet, kontinuerlig rennende nese, dårlig luktesans og ansiktsvondt eller hodepine og dette kan være veldig plagsomt. Det er økt forekomst av nesepolypper. Sinus-hypoplasi er vanlig og har høy spesifisitet for en PCD-diagnose. De fleste har også kronisk otitt med effusjon og hørselstap (KOME) særlig hos de yngre pasientene. KOME pleier å persistere gjennom barnealder, men er mindre vanlig i voksenalder. Residiverende mellomørebetennelse særlig i spebarnsalder er vanlig og det er økt forekomst av trommehinneperforasjon med kronisk ottorhea hos barn med KOME. Imidlertid stilles diagnosen sjeldent hos spedbarn siden slike symptomer er vanlige i denne alderen uten at det foreligger alvorlig underliggende sykdom. Omtrent en tredjedel av pasientene har et raskt progressivt sykdomsforløp. Hos en tredjedel av barn diagnostisert i førskolealder, er lungefunksjonen mindre enn 80 % av forventet ved første måling og bronkiektasier kan allerede forekomme. Det har tidligere vært hevdetat diagnose i ung alder er fordelaktig og kan gi bedre mulighet forstabilisering av lungefunksjon, men lungefunksjonen hos noen pasienter vil svekkes uansett. Det er økt forekomst av gastroøsofageal refluks, skoliose og hydrocephalus hos PCD-pasienter. De fleste menn med PCD er infertile, avhengig av hvilket gen som er involvert mens det er økt risiko for svangerskap utenfor livmoren hos kvinner. Levetid er antatt å være normal, men det finnes enda ikke longitudinelle studier som kan verifisere dette. I enkelte tilfeller er det behov for lungetransplantasjon.
Diagnostikk og utredning
Å stille en PCD-diagnose er en kompleks og vanskelig prosess. European Respiratory Society (ERS) PCD Task Force, har nylig publisert diagnostiske retningslinjer hvor det er sterkt anbefalt at diagnostiske tester ved PCD gjøres vedsentre med spisskompetanse og videre at testeresultatene tolkes av spesialister med spisskompetanse i PCD. I de fleste land er diagnostisk utredning sentralisert; i UK for eksempel, som har ca 65 millioner innbyggere, finnes det kun 3 diagnostiske sentre og de er finansiert av staten. Det finnes ikke noen enkel gullstandard-test for PCD. Fire kriteria-definerte kliniske karakteristikker i kombinasjon kan predikere PCD hos barn og unge med stor sannsynlighet, nemlig uforklarlig respirasjonsbesvær og forlenget behov for supplerende oksygen hos terminfødte barn, tidlig debut av helårligvåt hoste, tidlig debut av helårlignesetetthet og situs inversus eller ambiguus. Enda mer nøyaktig er PICADAR-score men selv da vil man gå glipp av ca 14 % av PCD-pasienter: https://erj.ersjournals.com/content/47/4/1103. Ca 5 % av førskolebarn med kronisk våthoste har PCD og andre årsaker til kronisk våthoste, for eksempel cystisk fibrose, immunsvikt, tracheobronchomalasi, og pulmonal aspirasjon bør utelukkes. Pasienter utredes i henhold til en trinnvis diagnostisk algoritme som er beskrevet nedenfor.
- Nasal nitrogen oksid (nNO)-måling. nNO hos PCD-pasienter er ekstremt lav av ukjente årsaker. Standarder for måling av nNO uttrykt som produksjonsrate er blitt publisert og ”cut-off ”-verdier under 77 nl/min er valide i ulikesentre, analysatorer og populasjoner. Dette samsvarer med ca 250 parts per billion (ppb). Den negative prediktive verdien av nNO >77 nl/min er 99,1 %, men likevel er dette ikke en absolutt verdi som kan utelukke tilstanden. nNO måling er vanskelig hos barn yngre enn 5–6 år når ”tidal-breathing” teknikk istedenfor ”breath-hold” må brukes. ”Cut-off” verdier for ”tidal-breathing” metoden er ikke enda etablert.
- Høyhastighets video-analyse av cilie-slagfrekvens og -mønster. Cilier til undersøkelse får en ved nesebørstebiopsi av inferior concha når pasienten har vært virusinfeksjonsfri i minst 4 uker. Vurdering av cilie-slagmønster krever høy kompetanse og erfaring. Det anbefales ikke at slagfrekvens alene måles uten samtidig vurdering av slagmønster. For å utelukke sekundær dyskinesi som skyldes infeksjon eller betennelse, anbefales det å etablere luft-væske-grensesnitt-kultur av epitelcellene for å regenerere ciliene før gjentatt undersøkelse. Høyhastighets video analyse og cellekultur tilbys enda ikke i Norge.
Klikk på lenkene under for å se eksempler på forskjellige tilfeller av cilieslagfrekvens og -mønstre:
https://youtu.be/6fDXyin8eMA
Normal ciliary beating from above
https://youtu.be/6cyOLaz-gMI
Normal ciliary beating (HD)
https://youtu.be/iDN5pmkec_0
Circular ciliary beating secondary to central microtubular defect from above
https://youtu.be/8TIkmmwpY5Y
Normal ciliary beating (planar)
- Transmisjon elektronmikroskopi (TEM)-udersøkelse av cilie-ultrastruktur anbefales som en del av utredning hvor det er mistanke om PCD, til tross for at denne undersøkelsen vil være normal hos ca 30 % av PCD-pasienter. Hos normale individer uten PCD vil ca 10 % av ciliene ha patologiske funn og det kan derfor være utfordrende å bestemme om patologiske funn taler for PCD eller ikke. Minst 50–100 cilie-tverrsnitt fra multiple cilier bør undersøkes og tolkning av TEM funn krever betydelig spisskompetanse. Mindre erfaring med TEM er assosiert med både over- og under-diagnostisering av PCD og det anbefales på det sterkeste at TEM ikke utføres ved sentre som ikke har betydelig erfaring. Det skal etter hvert etableres europeiske retningslinjer for TEM-diagnostikk som vil inkludere et system for kvalitetssikring. Tegning av cilieultrastruktur vises under.
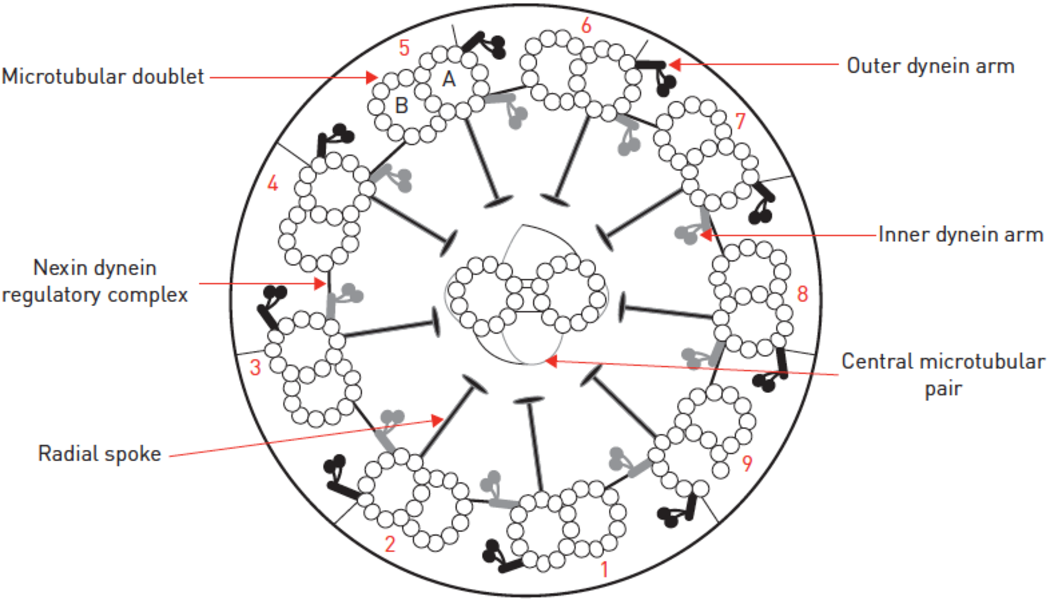
Figur er reprodusert fra Eur Respir J 2017;49:1601090
En metaanalyse av TEM funn fra 822 PCD-pasienter som representerer det største pasientmaterialet, viste at 44 % manglet ytre-dyneinarmer (YDA), 25 % indre-og ytre-dyneinarmer, 10 % indre-dyneinarmer (IDA) med mikrotubulære desorganisering (radial eik-defekt), 9 % indre-dyneinarmer, 8 % hadde sentral-par defekt og 1–3 % hadde cilieaplasi eller andre defekter. Cilier som mangler YDA eller YDA pluss IDA er statiske; de som mangler IDA eller IDA+radial eik defekt har et stivt slagmønster mens de som har sentral-par defekt har et sirkulært slagmønster. Visse genmutasjoner, for eksempel DNAH11, er assosiert med normal TEM, men ved bruk av 3D-rekonstruksjon (elektron-tomografi) kan subtile defekter påvises. I tilfellet DNAH11-mutasjoner ser man at YDA har ca 30 % mindre volum en vanlig. Vurdering av IDA er vanskelig også fordi cilier kan miste IDA som en konsekvens av viral infeksjon. Det anbefales derfor at TEM gjentas etter minimum 4–6 uker uten sykdom. Noen eksperter mener at mangel på IDA i seg selv, uten tilleggsfunn som mikrotubulær desorganisering, ikke forårsaker PCD. Det er derfor viktig at pasienter med isolerte indre dyneinarm-defekter ikke får diagnosen primær ciliær dyskinesi uten at det foreligger flere funn som støtter dette. - Genetikk. Det finnes mutasjoner i minst 36 gener som kan forårsake PCD, men det er mer enn 200 gener assosiert med bevegelige cilier. Hos populasjoner med en bekreftet eller høy sannsynlig PCD er årsaken genetisk betinget hos 50–80 % av pasientene. Sensitiviteten av genetisk testing som førstelinje-undersøkelse er ukjent. Mutasjoner i DNAH5 (dynein axomenal heavy chain og DNAI1-gener (dynein axomenal intermediate chaín 1 gener) står for 50–70 % av tilfellene med mangel på YDA. Nesten alle med mangel på IDA og mikrotubulær desorganisering vil ha mutasjoner i CCDC39 eller CCDC40-gener. Ca 20 % av individer med normal TEM vil ha mutasjoner i DNAH11-genet (dynein axomenal heavy chain 11). Mutasjoner i RGMC (reduced regneration of multiple cilia) genene CCNO og MCIDAS og i gener som koder for radiale eiker-proteiner (RSPH1, RSPH3, RSPH4a, RSPH9) er ikke assosiert med situs inversus eller ambiguus.
- Immunofluorescens. Det finnes antistoffer mot flere cilieproteiner inkludert antistoffer som er rettet mot YDA, IDA, radiale eik-hode, og dynein regulerende kompleks proteiner. Disse antistoffene med sekundære fluorescerende merker lokaliserer seg til cilieproteiner og kan visualiseres ved bruk av confocal eller fluorescerende mikroskopi. Mislokalisering av eller mangel på proteiner kan da påvises. Den første studien om bruk av immunofluorescens som diagnostisk verktøy ble publisert i februar 2017 og viste at sensitiviteten av undersøkelsen er for lav til å brukes som eneste test. Testen tilbys ikke enda i Norge.
Basert på disse 5 testene har ERS Task Force uttalt at pasienter med typiske symptomer på PCD samt enten positive TEM-funn eller ikke-tvetydige biallelic-mutasjoner har en sikker PCD diagnose. For pasienter med typiske PCD symptomer hvor det ikke er mulighet for genetiske eller TEM undersøkelser, kan veldig lav nNO pluss positive funn med høyhastighets videoanalyse ved minst 3 anledninger gi en høy sannsynlighet for en positiv PCD-diagnose. I Norge er en PCD-diagnose basert på typiske symptomer og kliniske funn, lav nNO og positive funn ved TEM i kombinasjon med genetikk.
Videre utredning
Alle barn med heterotaxi eller situs inversus bør utredes med ultralyd av hjerte og abdomen for å se etter hjertefeil og plassering av organer i abdomen, særlig med tanke på manglende milt.
Ved rask økning i hodeomkrets bør man tenke på hydrocephalus og bestille enten ultralyd eller MR caput.
Noen nyfødte barn har en vedvarende tachypnoea og bør henvises til utredning for å utelukke tracheobronchomalasi som ser ut til å være mer vanlig enn normalt hos barn med PCD.
Det er lav terskel for å gjøre 24-timers pH-måling gitt at PCD-pasienter har økt forekomst av gastroøsofageal refluks.
Bruk av metacholine-provokasjon som diagnostisk test for astma hos PCD-pasienter er ikke etablert og dessuten kan en positiv PD20 indikere ikke-spesifikk luftveisbetennelse som ikke vil respondere på inhalasjonssteroid behandling. FeNO pleier å være lavere enn normalt hos PCD-pasienter og kan ikke brukes for å utelukke astma.
Det er økt forekomst av D-vitamin-mangel og lav KMI før pubertet hos PCD-pasienter; årsaken til dette er ukjent.
Behandling
Det finnes nesten ingen klinisk forskning om hvordan PCD-pasienter bør behandles. De behandles som cystisk fibrose (CF)-pasienter uten at det finnes evidens for at dette er effektivt. For å imøtekomme mangelen på klinisk forskning om sykdommen er det etablert et Europeisk konsortium BEAT-PCD som er COST aksjon (Cooperation in Science and Technology) hvor Norge er representert. Gjennom dette fellesskapet kan internasjonalt forskningssamarbeid på sykdommen ivaretas og utvikles.
Lunger
Slimmobilisering er svært viktig for PCD-pasienter og er enbærebjelke i behandlingen. Fordi ciliene ikke fungerer er pasientene avhenging av fysioterapi i form av pusteøvelser (Active Cycle of Breathing), bruk av PEP-maske eller intermitterende CPAP, ”flutter” eller perkusjon sammen med hoste og særlig fysisk aktivitet for å få opp slim og redusere bakteriemengden.
Slimløsende behandling. Bruk av hypertont-saltvannsinhalasjoner (3–7 %) er etablert hos CF-pasienter, men dette er ikke undersøkt i kliniske studier hos PCD-barn. CF-pasienter har bedre lungefunksjon og mindre infektive ekaserbasjoner ved bruk av 7 % NaCl inhalasjoner og anekdotisk får noen PCD-pasienter god effekt av hypertone NaCl-inhalasjoner både mht slimmobilisering og lungefunksjon målt før og etter behandling er etablert. Hos mindre barn og særlig i det første leveåret kan man evaluere effekten av 0,9 eller 3 % NaCl-inhalasjoner. Sterkere 7 % NaCl kan forårsake bronkokonstriksjon, som kan unngås med å gi 0,1–0,4 mg salbutamol på kolbe før NaCl inhalasjon. Det finnes enkelte tilfeller hvor rhDNase (Pulmozyme) har vært effektivt hos PCD-pasienter med atelektase, men det er også beskrevet tilfeller av forverring. Det finnes ingen kliniske studier om rhDNase hos PCD-pasienter og bruk av den er ikke anbefalt. Det finnes en studie hos CF- og PCD-pasienter hvor oral N-acetylecystein (Mucomyst) var assosiert med bedre lungefunksjon hos CF-pasienter, men ikke hos PCD-pasienter. Oral N-acetylcystein anbefales derfor ikke. Det finnes ikke kliniske studier om N-acetylcystein-inhalasjoner. Mannitolinhalasjoner er assosiert med lengre tid til første eksaserbasjon og bedre livskvalitet hos CF-pasienter, men det finnes ingen studier av PCD-pasienter. Hos ikke-CF-bronkiektasi-pasienter er mannitolinhalasjoner assosiert med bedre livskvalitet og mannitolinhalasjoner kan vurderes på ”case-by-case” basis hos PCD-pasienter, men brukes ikke enda i Norge.
Anti-inflammatorisk behandling. Inhalasjonssteroider er uten effekt hos PCD-pasienter og bør unngås dersom pasienten ikke har astma som en tilleggsdiagnose. Salbutamol kan brukes før hypertont NaCl-inhalasjoner som nevnes over, men ellers finnes det ikke indikasjon for bronkodilaterende medikamenter hvis pasienten ikke har astma. Det finnes ingen rolle for bruk av systemiske steroider hos PCD-pasienter dersom de ikke har astma eller allergisk bronkopulmonal aspergillose (ABPA). Det er underveis en dobbel-blind randomisert kontrollert europeisk studie om sikkerhet og effekt av langtidsbehandling (6 måneder) med azitromycin hos PCD-barn og -voksne basert på tidligere erfaring med CF-pasienter.
Per oral antibiotika behandling i minst 2 uker basert på tidligere ekspektorat- eller larynksaspirat-dyrkning samt intensivering av slimmobilisering er anbefalt tidlig ved virale luftveisinfeksjoner eller økt slimdannelse og hoste. Ved dårlig allmenntilstand, inkludert feber og pustebesvær eller fortetninger på rtg thoraks eller manglende effekt av per oral antibiotika behandling, kan det være indikasjon for intravenøs antibiotikabehandling, vanligvis i 10–14 dager. Ca 25 % av PCD-pasienter innlagt på sykehus til iv antibiotikabehandling pga forverring har vedvarende lavere spriometri-verdier etter utskrivelse. De vanligste luftveismikrobene hos PCD-pasienter er H. Influenzae (ikke-typebestembar), S. pneumoniae, M. cattarhalis og S aureus. Forekomsten av infeksjon med Ps. Aeruginosa er mindre hos PCD-pasienter enn CF-pasienter, men behandles på samme måten (se kapittel Cystisk fibrose) både ved akutt og kronisk infeksjon. Det er viktig å være oppmerksom på at man bruker en høyere dose av iv aminoglykosider hos CF-pasienter. Det finnes ikke rapporterte tilfeller hvor det har vært kryssinfeksjon ved Ps. aeruginosa mellom PCD-pasienter, men inntil videre bør likevel de samme infeksjonskontrollreglene brukes som ved CF. Det er økt risiko for ikke-TB mykobakterie-infeksjon ved langtidsbruk av azitromycin. Mange PCD-pasientene får ikke feber eller utslag på CRP ved akutte forverringer og disse undersøkelsene skal ikke brukes for å bestemme om en pasient skal behandles med antibiotika eller ikke. PCD-pasienter får lett atelektase særlig ved akutte forverringer, slik at man bør ha lav terskel for å gjøre rtg thoraks. Alle pasienter med akutt atelektase bør få antibiotikabehandling og intensivering av slimmobilisering. Snarlig bronkoskopi anbefales ved akutt atelektase og bør vurderes etter kontakt med PCD-kompetansesenteret.
Vaksinasjon
I tillegg til vanlig barndomsvaksinasjoner er det anbefalt vaksinasjon mot influensa for barn >6 måneder gamle. Alle, og særlig de som ikke har milt, bør vaksineres mot pneumokokkinfeksjon. I motsetning til barn med CF anbefales RS-virus-vaksine det første leveåret for PCD-barn, særlig når pasienten har vært alvorlig syk etter fødsel og ved komorbiditet, for eksempel tracheobronkomalasi som anedotisk har økt forekomst hos yngre PCD-barn.
Øvre luftveier
Kronisk rhinosinusitt. Målet for behandlingen er å redusere nesetetthet, fjerne purulent sekret og forbedre luktesans og livskvalitet. En gang daglig eller hyppigere neseskylling med 0.9 % eller sterkere 3 % NaCl anbefales ved bruk av dråper, nesehorn eller neseforstøver. Noen pasienter mener at å skylle nesen er den mest effektive og behagelige formen for PCD-behandling. Pasienter med polypper behandles ofte med nasale steroider uten at det finnes evidens for det hos PCD-pasienter. For eldre barn og voksne som er veldig plaget av nesetetthet eller sinusitt kan FESS (Functional Endoscopic Sinus Surgery) være en effektiv løsning. FESS åpner et større hull mellom nesen og sinusen som gir bedre tilgang for inhalasjoner og bedre drenasje av sekret.
Kronisk otitt med effusjon (KOME). Mange unge barn med PCD har hørselstap som skyldes væske i mellomøret og behandlingen av disse pasientene er kontroversiell. Behandling bør vurderes ved hørselstap >25 dB i mer enn 3 måneder. Bruk av trommehinnedren er assosiert med 10–50 % forekomst av permanent trommehinneperforasjon og vedvarende lekkasje uten at det nødvendigvis gir en forbedring i hørselen. I noen få tilfeller kan man prøve med innleggelse av dren i ett øre, ikke to, men de fleste barn behandles med høreapparat. Det er sterkt behov for kliniske studier om hvordan hørselstap best kan behandles.
Anbefalinger om oppfølging
PCD-pasienter bør følges opp regelmessig av leger med kompetanse innenfor PCD.
Lunger
Unge pasienter bør de første leveårene kontrolleres av lungelege hver 1-2 måneder avhenging av klinisk tilstand og deretter minst hver tredje måned. Som ved CF-pasienter bør det gjøres årlige kontroller. Ved hver kontroll og ved forverring bør det tas larynksaspirat eller ekspektorat til bakteriedyrkning, inkludert CF-patogener. Det bør også sjekkes for sopp og virus ved PCR. Lungefunksjon kontrolleres ved hver klinikkvisitt, helst før og etter slimmobilsering, som kan føre til en økning i FEV1 inntil ca 30 %. Hvis mulig bør en fysioterapeut være tilgjengelig for alle pasienter ved hver poliklinisk visitt og tilgjengelig på telefon innimellom visittene. Barnehage- og skolepersonale bør få opplæring om PCD og viktigheten av aktivitet for god slimmobilisering. En spesialist PCD-sykepleier som kan ringes i vanlig arbeidstid eller få beskjed på telefonsvarer, bør være tilgjengelig.
Øvre luftveier
Minst årlige kontroller hos ØNH-lege med PCD kompetanse.Hørselstest minst hver 6 måned i barndomsalder og hyppigere ved behov.
Vanlige kontroller
Grundig sykehistorie om symptomer fra øvre og nedre luftveier, inkludert ører og hørsel, antall forverringer, antibiotikabruk, slimmobilsering og bruk av inhalasjoner, aktivitetstoleranse, søvnkvalitet, matlyst og gastroøsofageal refluks.
Kliniske undersøkelser som inkluder inspeksjon av ører og nese samt lungeundersøkelse
SpO2, respirasjonsfrekvens, høyde, vekt
Lungefunksjon (tidalflow volum, spirometri, plethysmografi) før og etter slimmobilisering hvis mulig
Ekspektorat/larynksaspirat dyrkning (vanlig pluss CF patogener), sopp og virus PCR
Nesesekret-dyrkning.
Årlige kontroller
Som over
Ergospirometri
Rtg thoraks
CT thoraks gjøres en eller to ganger i løpet av barndomsalder
Blodprøver: Hb, hvite, diff, trombocytter, elektrolytter, urea, kreatinin, leverfunksjon, total IgE, aspergillus spes IgE, IgG, IgA, IgM, D-vitamin.
Ekspektorat/larynksaspirat som over, pluss ikke-TB mykobakterie-dyrkning
Time hos spesialistsykepleier (livskvalitetsskjema pluss opplæring)
Time hos fysioterapeut
Time hos ØNH-lege
Time hos ernæringsfysiolog (i tilfellet D-vitaminmangel eller lav KMI)
Time hos sosionom (etter behov)
Time hos psykolog (etter behov)
PCD-kompentansesenter og omsorg for PCD-pasienter
OUS er blitt et kompetansesenter for PCD og som en del av denne tjenesten er det opprettet et nasjonalt register og biobank. Dette gir mulighet for kvalitetssikring og forskning. Det er ventet at omsorg for PCD-pasienter vil følge CF-modellen hvor ansvar for pasientoppfølging deles mellom kompetansesenteret og lokalsykehus og årlige kontroller skjer på kompetansesenteret. Diagnostiske tjenester bør av kapasitets- og kompetansemessige grunner være sentralisert og utvikles i henhold til anerkjente internasjonale retningslinjer. Dermed får man samlet den diagnostiske kompetansen og samtidig lagt til rette for formidling av kunnskap og erfaring til samarbeidspartnere ved andre behandlingsinstitusjoner.
Referanser
- Up to Date. Primary Ciliary Dyskinesia. Updated 13.02.17
- Leigh MW, Ferkol TW, Davis SD et al. Clinical features and associated likelihood of primary ciliary dyskinesia in children and adolsecents. Am Ann Thorac Soc 2016;13:1305-1313.
- Goutaki M, Meier AB, Halbeisen FS et al. Clinical manifestations in primary ciliary dyskinesia: systematic review and meta-analysis. Eur Respir J 2014;48:1081-1095.
- Kouis P, Papatheodorou SI, Yiallouros PK. Diagnostic accuracy of nasal nitric oxide for establishing diagnosis of primary ciliary dyskinesia: a meta-analysis. BMC Pulmonary Medicine 2015;15:153.
- Lucas J, Barbato A, Collins SA et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J Published Online First 26 November 2016 doi: 10.1183/13993003.01090-2016
- Morgan LC, Birman CS. Impact of primary ciliary dyskinesia on the upper respiratory tract. Paediatr Respir Rev 2016;18:33-38.
- Behan L, Leigh MW, Dell SD et al. Validation of a health-related quality of life instrument for primary ciliary dyskinesia. Thorax Published Online First: 28 February 2017 doi:10.1136/thoraxjnl-2016-209356
- Polieni D, Davis SD, Dell SD. Treatment recommendations in primary ciliary dyskinesia. Paediatr Respir Rev 2016;18:39-45.
- Shoemark A, Frost E, Dixon M et al. Accuracy of immunofluorescence in the diagnosis of primary ciliary dyskinesia. AJRCCM 2017; in press. Published 15 February 2017.
- Behan L, Dimitrov BD, Kuehni C et al. PICADAR: a predictive diagnostic tool for primary ciliary dyskinesia. Eur Respir J 2016;47:1103-1112.