









Generell veileder i pediatri
8. Hjerte- og karsykdommer
8.24 Kardiomyopatier
Sist faglig oppdatert: 19.06.2025
Marit Kristine Smedsrud
Bakgrunn
Kardiomyopatier er primære hjertemuskelsykdommer som kjennetegnes av strukturelle, funksjonelle og elektriske forstyrrelser i hjertet. Det er en relativ heterogen sykdomsgruppe, hvor den kliniske presentasjonsformen kan variere betydelig hos pasienter med samme diagnose. Sykdommene kan debutere med tydelige strukturelle forandringer i hjertet, mens man i andre tilfeller ser ventrikulær arytmi og plutselig død før tydelige strukturelle forandringer er synlige.
Kardiomyopatier hos barn har svært varierende alvorlighetsgrad. En del barn utvikler alvorlig sykdom med tidlig debut, rask sykdomsprogresjon og dårlig prognose, men andre kun har lette myokardforandringer, ofte identifisert i forbindelse med familiescreening. Kardiomyopatier er en av sykdomsgruppene innenfor barnekardiologi med dårligst prognose. Nær 40 % av barn med symptomgivende kardiomyopati gjennomgår en hjertetransplantasjon eller dør innen 2 år etter at de har fått diagnosen (1).
I kardiomyopati-retningslinjene fra European Society of Cardiology (ESC) som ble presentert i 2023 (2) deles kardiomyopatiene inn i fem hovedfenotyper:
- Hypertrofisk kardiomyopati (HCM)
- Dilatert kardiomyopati (DCM)
- Arytmogen høyre ventrikkel-kardiomyopati (ARVC)
- Ikke-dilatert venstre ventrikkelkardiomyopati (NDLVC)
- Restriktiv kardiomyopati (RCM)
Diagnostikk og utredning
I anamnesen vektlegges klassiske hjerterelaterte symptomer som tungpust, redusert utholdenhet, hjertebank, svimmelhet, synkope og nærsynkope, samt takypné, dårlig vektoppgang og svetting ved amming hos de yngste. Ved diagnostikk av genetiske hjertesykdommmer er særlig synkope og nærsynkope, og om dette har skjedd i relasjon til fysisk aktivitet, viktig anamnestisk informasjon. Videre er familieanamnesen sentral, med særlig fokus på kjente hjertesykdommer og plutselig død i ung alder (inkludert plutselig død av antatt andre årsaker, f.eks. uforklarlige drukninger og ulykker).
Ved kortvarig sykehistorie og akutt symptomdebut er myokarditt ofte en differensialdiagnose. Det bør da tas serologi, og ev. PCR i blod, for bl.a. parvovirus, adenovirus, EBV, CMV, HSV, HHV 6–8, influensa A og B, mykoplasma og enterovirus.
Hos nyfødte er det i anamnesen viktig å legge vekt på mulige toksiner, infeksjoner, legemidler eller hjerterytmeforstyrrelser hos fosteret under svangerskapet. Patologiske urinprøver kan peke i retning av bakenforliggende syndromer eller metabolske sykdommer (ketoner, aminosyrer, organiske syrer, oligosakkarider, mukopolysakkarider). Det finnes en rekke aktuelle blodprøver i differensialdiagnostikken. Initialt bør det hos alle nyfødte som utredes for kardiomyopati tas følgende laboratorieprøver: Hb, leukocytter med differensialtelling, trombocytter, albumin, aminosyrer, ammoniakk, proBNP, karnitin, kalsium, kolesterol, ceruloplasmin, glukose, laktat, leverparametre, magnesium, pyruvat, selen, thyroideaparametre, vitaminer (D, B1, C), ANA, anti Ro/La, senkning, CRP og troponin.
Ekkokardiografi er generelt det viktigste verktøyet for å diagnostisere kardiomyopati. Når det gjelder utregning av z-score (standardavvik fra det forventede gjennomsnittet), anbefaler ESC-retningslinjene å bruke normative data fra Pediatric Heart Network (3). Z-score må imidlertid brukes med forsiktighet hos små barn, hvor feilmargin kan gi større endringer i z-score og hos barn hvor kroppsoverflaten er utenfor 2 SD for alder. Det er indikasjon for arbeids-ekko hos HCM-pasienter med anstrengelsesrelaterte symptomer uten signifikant utløpsobstruksjon i hvile.
12-kanalers EKG er sentralt i utredningen. Påfallende hjertefrekvens, unormal AV-overledning og avvikende hjerteakse, QRS-morfologi, -spenning og -varighet, samt forstyrrelser i repolariseringstid kan være et signal om en bakenforliggende arvelig hjertesykdom. Senpotensial-EKG brukes ved ARVC. Arbeids-EKG er ofte nyttig både for å fremprovosere anstrengelsesutløste arytmier, vurdere arbeidskapasitet og puls- og BT-respons, og for kvalifiserte vurderinger vedrørende treningsråd. I tillegg er langtids EKG-registreringer meget viktige i oppfølgingen av genetiske hjertesykdom.
Magnet-resonans (MR) av hjertet er bedre enn ekko når det gjelder å diagnostisere apikal hypertrofi, aneurismer (opptil 40 % oversees ved ekko) og tromber. MR med kontrast gir også informasjon om sen kontrastoppladning (late gadolineum enhancement, LGE) som uttrykk for fibrose. LGE er en prognostisk markør for dårligere prognose og bl.a. for økt arytmitendens. MR av hjertet utføres som ledd i utredningen hos pasienter med myokardhypertrofi og bør også vurderes gjennomført hos personer med normal hjertefunksjon dersom de er bærere av sykdomsfremkallende varianter av gener forbundet med økt risiko for livstruende arytmier (f.eks. FLNC, DES, DSP, PLN, LMNA, TMEM4 og RMB20).
Hypertrofisk kardiomyopati
Ved hypertrofisk kardiomyopati (HCM) ses venstre ventrikkelhypertrofi uten at det kan forklares av vanligere tilstander som høyt blodtrykk eller klaffefeil.
Diagnostikk og utredning
For å sette diagnosen hos barn kreves det en veggtykkelse på mer enn 2 standardavvik over det forventede gjennomsnittet (z-score > 2). Veggtykkelsen er ofte asymmetrisk og gjerne mest uttalt i ventrikkelseptum. Det kan gi obstruksjon av venstre ventrikkels utløpstrakt (LVOTO) med varierende grad av systolisk fremoverrettet bevegelse av mitralklaffen (systolic anterior motion; SAM).
Prevalens og genetikk
HCM er den nest vanligste årsaken til kardiomyopati hos barn. Etiologien ved HCM hos barn er mer heterogen enn den man ser i den voksne populasjonen og inkluderer genetiske syndromer (f.eks. Noonan syndrom), nevromuskulære sykdommer (f.eks. Friedreichs ataksi) og avleirings-/metabolske sykdommer (f.eks. Pompe sykdom). De fleste tilfellene er imidlertid forårsaket av varianter i hjertesarkomerprotein-genene og er autosomalt dominant nedarvet. Den relative forekomsten av forskjellige HCM-etiologier varierer med alder: HCM relatert til metabolsk sykdom og genetiske syndromer diagnostiseres oftest i løpet av de første to årene av livet og har da ofte en dårlig prognose, mens HCM på bakgrunn av nevromuskulære lidelser oftest presenterer seg i ungdomsårene. Historisk sett ble sarkomerisk sykdom ansett som uvanlig hos barn, men nyere europeiske og nord-amerikanske studier har vist at sarkomerisk sykdom også kan debutere i tidlige barneår (3,4). Disse studiene viser at flertallet av dem som blir diagnostisert med HCM gjennom familiescreening, oppfyller diagnostiske kriterier før de blir tenåringer, med en ikke ubetydelig andel diagnostisert i spedbarnsalder. Familiær sarkomerisk HCM er assosiert med variabel penetrans. Mens noen individer utvikler hypertrofi i tidlig barndom, vil andre variantbærere aldri utvikle sykdom. Sykdomsfenotype og -progresjon er også svært varierende, selv blant familiemedlemmer som bærer en identisk genvariant (5). Debut i barnealder er mer sannsynlig dersom det er andre tilfeller med tidlig debuterende hjertemuskelsykdom i familien (6).
Symptomer og funn
En stor andel av pasientene med HCM er asymptomatiske, mens andre kan ha uttalte symptomer og utvikle livstruende arytmier og hjertesvikt. De vanligste symptomene er dyspné, palpitasjoner og svimmelhet, samt synkope/nærsynkope og hjertestans. Ved utløpsobstruksjon er symptomene ofte anstrengelsesrelaterte. Reduksjon av venstre ventrikkels ejeksjonsfraksjon (LVEF) ses sjelden ved sarkomerisk sykdom i barnealder. Strainanalyser kan imidlertid identifisere regional myokarddysfunksjon, typisk lokalisert i området med maksimal hypertrofi, og langaksefunksjonen er ofte svekket, selv hos dem med hyperdynamisk systolisk funksjon (7). Diastolisk dysfunksjon foreligger ofte ved etablert sykdom og kan til og med ses før utviklingen av hypertrofi. Hjertesviktrelaterte dødsfall dominerer i spedbarnsalderen, mens plutselig hjertedød er den vanligste dødsårsaken utenom spedbarnsalder med en rate på 1–2 % per år (8). Dette er 50 % høyere enn det som er rapportert i voksne HCM-populasjoner. Plutselig hjertedød ser ut til å være svært sjelden under 6 års alder (8).
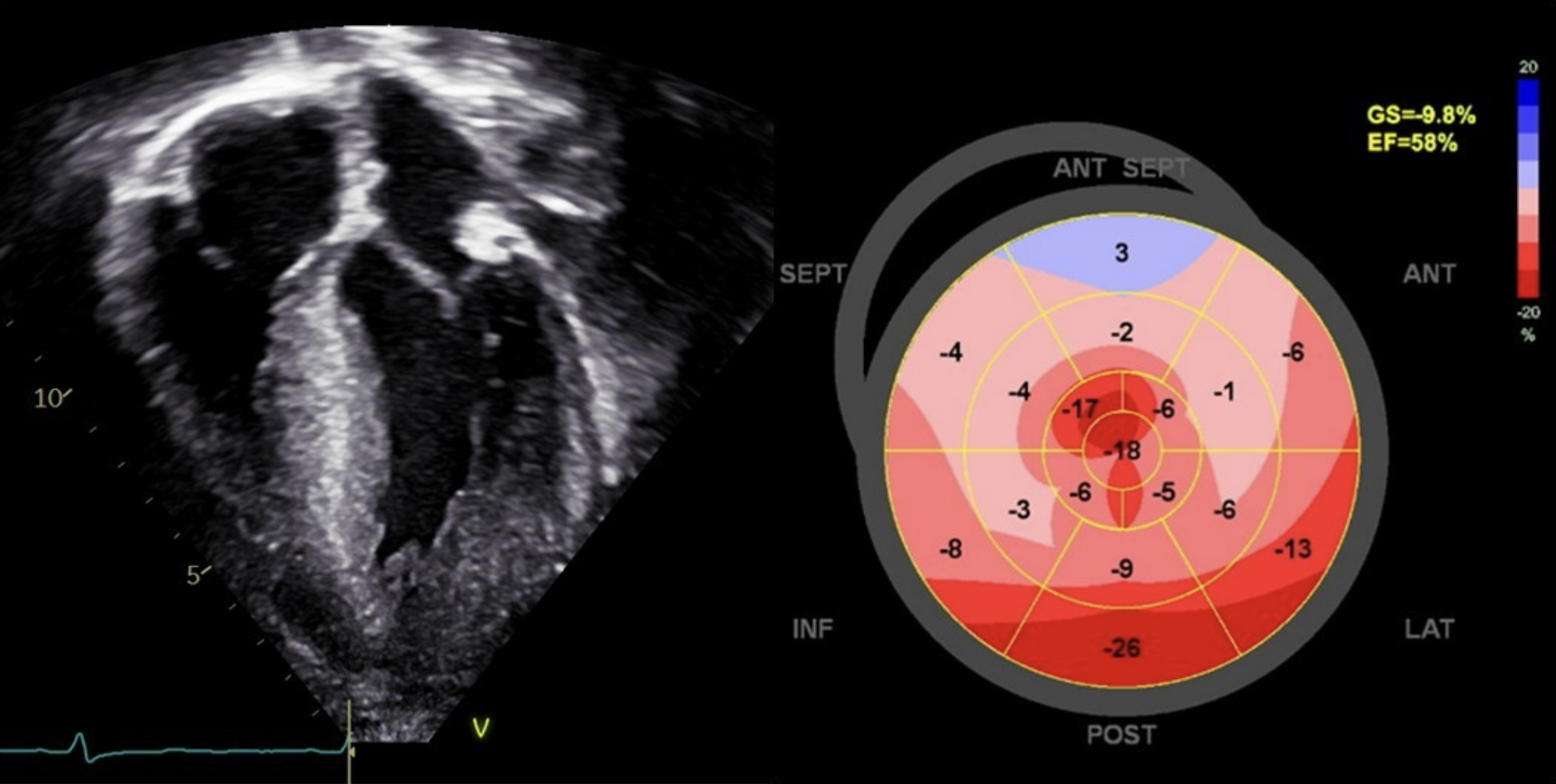
Behandling og oppfølging
For HCM-pasienter med hjertesviktsymptomer på tross av bevart LVEF, rettes behandlingen mot diastolisk dysfunksjon, først og fremst ved å redusere hjertefrekvensen. Førstehåndspreparat for hjertefrekvensreduksjon er betablokker i høyeste tolererte dose. Hos HCM-pasienter med redusert LVEF anbefales ordinær hjertesviktbehandling, men man skal være forsiktig med diuretikabehandling og vasodilatasjon hos pasienter med uttalt hypertrofi og ved utløpsobstruksjon. Hos symptomatiske pasienter med LVOTO er målet å redusere symptomene ved bruk av medikamenter eller septumreduserende intervensjon. Medikamentell behandling rettet mot obstruksjonen i utløpstraktus er først og fremst høyeste tolerable betablokker-dose. For symptomatiske pasienter med hvile- og/eller provosert LVOTO ≥ 50 mm Hg til tross for maksimal medikamentell behandling vurderes septumreduserende behandling. I henhold til ESC-retningslinjene anbefales septal myektomi fremfor septumablasjon med alkohol hos barn (2). Mavakamten (myosinhemmer) brukes for behandling av hos voksne med symptomatisk HOCM, men er foreløpig ikke godkjent til barn under 18 år. Det foregår imidlertid studier.
Forebygging av plutselig hjertedød
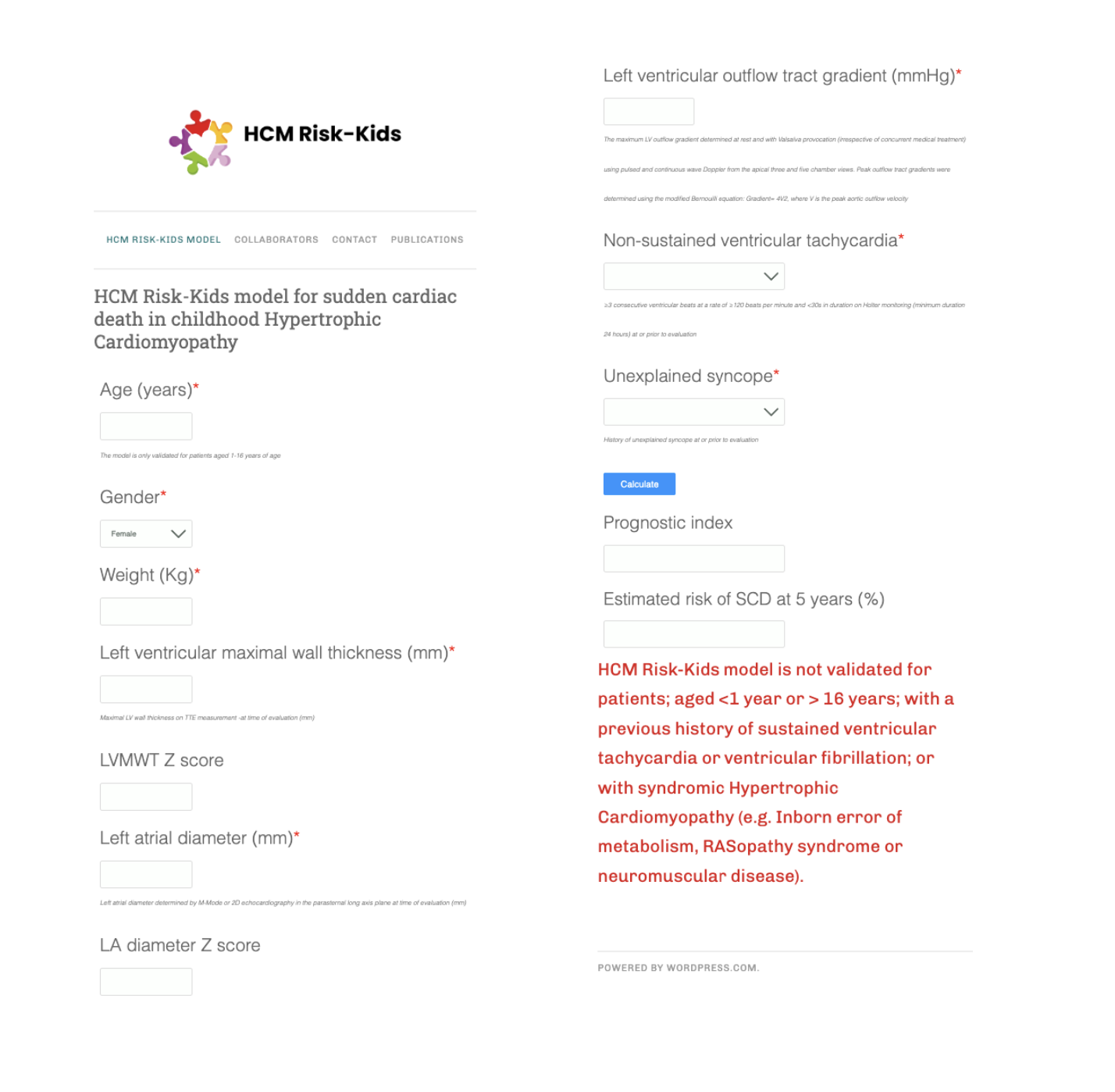
Når det gjelder forebygging av plutselig hjertedød, er det ikke vitenskapelig bevist at betablokker virker forebyggende. Å vurdere om det foreligger indikasjon for primærprofylaktisk ICD er utfordrende og forblir en klinisk vurdering. Det finnes ingen randomiserte studier eller statistisk validerte prospektive prediksjonsmodeller som kan brukes til å veilede ICD-implantasjon, men i ESC-retningslinjene anbefales det å bruke HCM-risikokalkulator som et hjelpemiddel for å vurdere risiko for potensielt livstruende ventrikulære arytmier. For barn under 16 år finnes det to konkurrerende kalkulatorer som har vist ganske lik treffsikkerhet (HCM Risk-Kids model for sudden cardiac death in childhood og PRIMaCY Childhood HCM Sudden Cardiac Death Risk Prediction tool). Ingen av disse er validert for barn med syndromal HCM eller med HCM relatert til metabolsk sykdom.
Konsekvenser for idrett
Tidligere retningslinjer har anbefalt at pasienter med HCM og høy risiko for ventrikulære arytmier og plutselig hjertedød skal avstå fra konkurranseidrett. Det foreligger imidlertid nyere forskning som skaper tvil om høyintensitetsaktivitet virkelig er uheldig hos HCM-pasienter. Idrettsutøvere med mild venstre ventrikkel-hypertrofi, normal diastolisk funksjon og ingen LVOTO er ofte i stand til å trene intensivt. Genotype-positive/fenotype-negative pasienter kan delta i all konkurranseidrett, men det anbefales årlig kardiologisk vurdering av disse (2). En av utfordringene i oppfølgingen er å skille fysiologisk venstre ventrikkelhypertrofi fra patologi hos idrettsutøvere. Evaluering av septumtykkelse-progresjon samt volum av venstre ventrikkel er viktig for å skille treningsindusert myokardhypertrofi fra tidlig HCM-sykdom hos ungdom (9).
Familiescreening
I henhold til oppdaterte retningslinjer anbefales det å starte screening av asymptomatiske familiemedlemmer, inkludert barn, på det tidspunktet HCM er diagnostisert i familien (2). Ved prediktiv gentesting av barn under 16 år skal begge foreldrene ha genetisk veiledning og samtykke til testingen.
Dilatert kardiomyopati
Dilatert kardiomyopati (DCM) er en heterogen sykdomsgruppe karakterisert av dilatasjon og redusert systolisk funksjon av venstre ventrikkel.
Diagnostikk og utredning
Venstre ventrikkeldilatasjon er definert ved endediastolisk dimensjon eller volum > 2 standardavvik over det forventede gjennomsnittet (z-score > 2). Redusert systolisk funksjon av venstre ventrikkel er definert ved LVEF < 50 % (2).
Prevalens og genetikk
DCM er den vanligste årsaken til kardiomyopati i barndommen og er forbundet med stor risiko for død. Viktige årsaker til DCM i barndommen er gjennomgått myokarditt, takyarytmier, toksiner (inkludert kjemoterapi), familiær DCM, medfødte metabolske sykdommer og nevromuskulære sykdommer. Man finner holdepunkter for myokarditt hos en stor andel av barn med DCM som gjennomgår tidlig endomyokardbiopsi, og forskning viser at personer med varianter i gener som koder for strukturelle proteiner i hjertet, kan være spesielt utsatt for alvorlig myokarditt (10). Familiær DCM er arvelig, og antallet kjente sykdomsfremkallende genvarianter øker. Noen eksempler er lamin A/C (LMNA), titin (TTN), fosfolamban (PLN), RNA-binding motif-protein (RBM20), Bcl-2-assoisiert athanogen (BAG) og filamin C (FLNC). Fenotypen varierer mellom de ulike genene.
Symptomer og funn
Barn med DCM kan debutere med hjertesvikt, med symptomer som varierer fra spisevansker til kardiovaskulær kollaps. Etter spedbarnsalder er DCM den vanligste årsaken til hjertetransplantasjon hos barn (1). Sykdommen kan også debutere med arytmier. Dette gjelder særlig for pasienter med LMNA-varianter hvor atrieflimmer, AV-blokk og livstruende ventrikulære arytmier ses relativt hyppig. Noen av disse pasientene har i tillegg skjelettmuskeldystrofi (f.eks. Emery-Dreifuss muskeldystrofi).
Behandling og oppfølging
Hos de fleste pasienter med genetisk betinget DCM gjelder vanlig hjertesviktbehandling (se kapittel om hjertesvikt hos barn i generell veileder). Behandlingsanbefalinger for asymptomatisk venstre ventrikkeldysfunksjon eller -dilatasjon er mangelvare, noe som gir en utfordring ved genetisk DCM, hvor en betydelig andel av pasientene er barn uten eller med milde symptomer, ofte fanget opp ved familiescreening. Ved DCM relatert til varianter i LMNA, troponin T (TNT), FLNC, desmin (DES), desmoplakin (DSP), PLN, transmembran protein 43 (TMEM43) og RMB20 må man ha fokus på symptomer og tegn på arytmi.
Forebygging av plutselig hjertedød
Risikostratifisering med tanke på plutselig hjertedød er en stor utfordring når det gjelder pasienter som er bærere av sykdomsfremkallende genvarianter som disponerer for DCM. ICD er effektivt for å forhindre hjertestans, men er også forbundet med komplikasjoner, spesielt hos unge pasienter, som vil trenge utskiftninger av elektroder så lenge de er i vekst. Pasienter med sykdomsfremkallende varianter i PLN, DSP, LMNA, FLNC, TMEM43 og RBM20 er vist å ha en betydelig høyere frekvens av alvorlige arytmiske hendelser uavhengig av LVEF (2) og man bør vurdere MR-undersøkelse (med LGE) hos disse selv om hjertefunksjonen er normal ved ekko cor.
Konsekvenser for idrett
Studier på voksne pasienter med DCM viser at moderat trening forbedrer fysisk kapasitet og ventrikkelfunksjon. Genpositive barn kan delta i fysisk aktivitet inntil hva de selv orker, men de skal ikke presses utover det. Høyintensitetsidretter og en idrettskarriere anbefales i utgangspunktet ikke. Intensiv trening og konkurranseidrett kan utløse dødelige arytmier, og personer med sykdomsfremkallende varianter av gener forbundet med økt risiko for livstruende arytmier bør vise varsomhet. Å avstå fra konkurranseidrett er særlig viktig for pasienter med sykdomsfremkallende varianter i LMNA, da det er data som tyder på at trening kan ha en negativ effekt på hjertefunksjonen hos disse (11).
Familiescreening
Det anbefales å starte screening av asymptomatiske familiemedlemmer, inkludert barn, på det tidspunktet DCM er diagnostisert i familien. Oppfølgingen individualiseres i forhold til ev. forekomst av tidlig sykdomsdebut i familien og forventet utvikling av sykdommen (2).
Ikke-dilatert venstre ventrikkelkardiomyopati
Ikke-dilatert venstre ventrikkelkardiomyopati (NDLVC) er en ny hovedfenotype i 2023-retningslinjene fra ESC (2). NDLVC defineres som en ikke-dilatert venstre ventrikkel med fettinfiltrasjon eller fibrose som ikke er relatert til iskemi, uavhengig av om systolisk funksjon er normal eller redusert. Betegnelsen kan også brukes i de tilfellene det foreligger isolert global hypokinesi uten arrdannelse (som ikke kan forklares av unormale loading-forhold eller koronarsykdom).
Diagnostikk og utredning
MR-undersøkelse av hjertet står sentralt i diagnostikken, og MR hjerte bør vurderes også hos personer med normal hjertefunksjon dersom de er bærere av sykdomsfremkallende varianter av gener forbundet med økt risiko for livstruende arytmier (f.eks. FLNC, DES, DSP, PLN, LMNA, TMEM4 og RMB20) (2).
Genetikk
De vanligste involverte sykdomsfremkallende genvariantene er DSP, FLNC, LMNA og PLN. Det er overlapp i den genetiske bakgrunnen for NDLCV, DCM og ARVC. NDLVC-fenotypen inkluderer pasienter som inntil nylig hadde diagnosen DCM uten å oppfylle kriterier for dilatasjon av venstre ventrikkel og pasienter med venstre-dominant ARVC.
Symptomer og funn
De fleste pasienter med NDLVC er asymptomatiske, men noen utvikler symptomer relatert til arytmi eller ledningssykdom (f.eks. synkope og hjertebank) eller diastolisk hjertesvikt (f.eks. dyspné). Ventrikulær arytmi, hjertestans eller plutselig hjertedød kan være det første tegnet på sykdommen hos noen av pasientene.
Behandling og oppfølging
Forebygging av plutselig hjertedød
Prediksjon og forebygging av plutselig hjertedød er hjørnesteinene i utredningen og oppfølgingen av pasienter med NDLVC. Som ved de andre kardiomyopati-undergruppene anbefales ICD-implantasjon hos overlevende etter hjertestans og hos pasienter som har opplevd livstruende ventrikulær arytmi. Data på pasienter med NDLVC er begrenset, men tilgjengelig forskning tyder på at genotype spiller en viktig rolle og at pasienter med varianter i PLN, TMEM43, DES, DSP, LMNA, FLNC (trunkerende varianter) og RBM20 har en betydelig høyere frekvens av alvorlige arytmi uavhengig av LVEF. Anbefalinger for primærforebyggende ICD-implantasjon er de samme ved NDLVC som ved DCM (2).
Konsekvenser for idrett
Anbefalinger for deltagelse i idrett er de samme ved NDLVC som ved DCM (se ovenfor) (2).
Familiescreening
Det anbefales å starte screening av asymptomatiske familiemedlemmer, inkludert barn, på det tidspunktet NDLVC er diagnostisert i familien (2). Oppfølgingen individualiseres i forhold til ev. forekomst av tidlig sykdomsdebut i familien og forventet utvikling av sykdommen. Jevnlige Holter-registreringer er særlig viktig hos pasienter med varianter av gener forbundet med økt risiko for livstruende arytmier.
Arytmogen høyre ventrikkel-kardiomyopati (ARVC)
ARVC er en myokardsykdom karakterisert av økt risiko for ventrikulære arytmier og karakteristiske strukturelle forandringer i hjertemuskelen, forårsaket av progressiv atrofi av hjertemuskelcellene som erstattes med fett og bindevev. Høyre hjertehalvdel er ofte mest affisert, men biventrikulær affeksjon er ikke uvanlig.
Diagnostikk og utredning
Diagnosekriteriene er komplekse og omfatter både bildediagnostikk, histologi, EKG, rytmeregistrering og familiehistorikk (12). For dem som er involvert i vurderingen av pediatriske pasienter med genvarianter som er forbundet med ARVC, er det imidlertid viktig å være klar over at diagnosekriteriene ble utviklet på bakgrunn av studier hvor gjennomsnittsalderen på pasientene var 38 år og kun 8 % av deltagerne var under 18 år. Prekordiale T-inversjoner er for eksempel ett av diagnosekriteriene, men dette teller ikke med hos barn under 14 år.
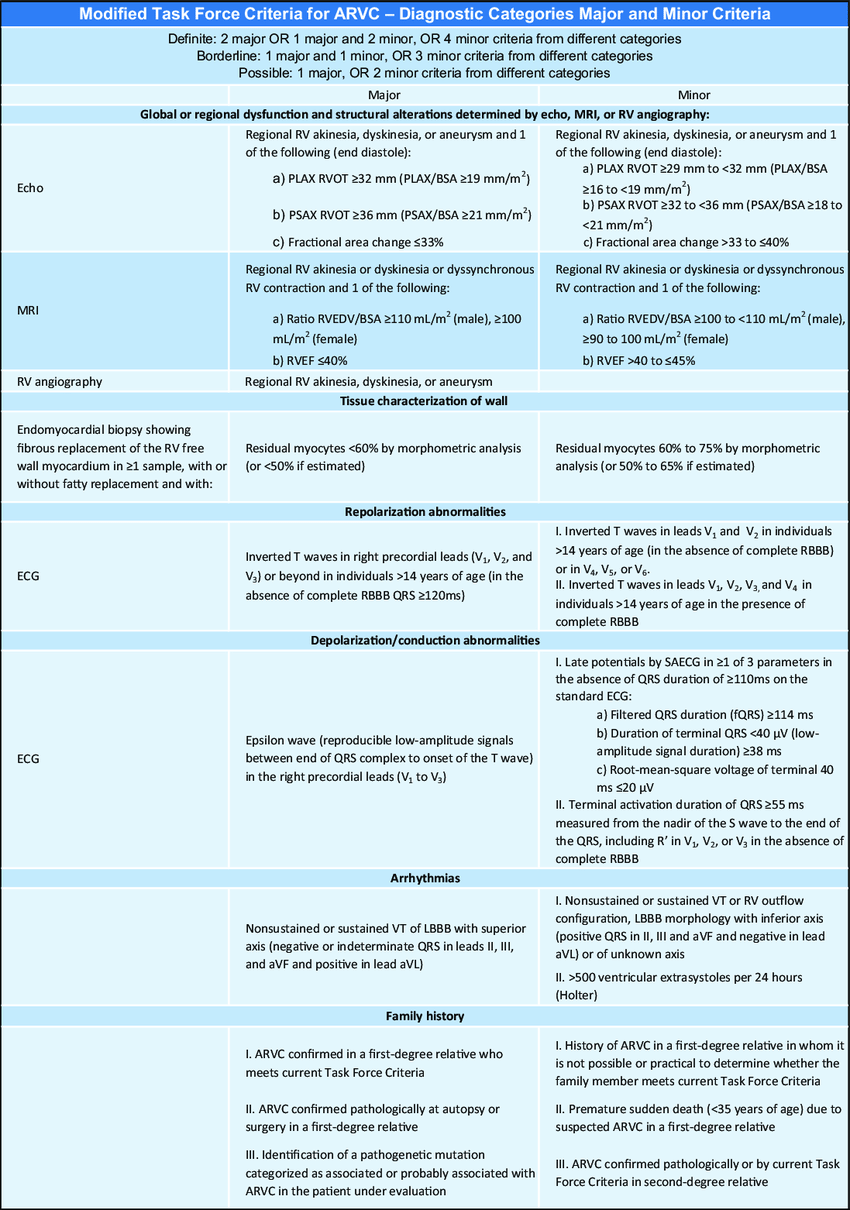
Genetikk
Genene som ligger til grunn for ARVC, koder hovedsakelig for proteiner i de kardiale desmosomene: plakofillin-2 (PKP2), desmoplakin (DSP), desmoglein-2 (DSG2), desmocollin-2 (DSC2) og plakoglobin (JUP). De kardiale desmosomene fungerer som «limet» mellom hjertemuskelcellene. Defekte desmosomer fører til at myocyttene «rives fra hverandre», nekrotiserer og erstattes av fett og bindevev når hjerteveggen utsettes for stort mekanisk stress. Arvegangen er hovedsakelig autosomal dominant.
Symptomer og funn
En ARVC-diagnose bør mistankes ved synkope eller hjertestans relatert til fysisk aktivitet. Hyppige ventrikulære ekstraslag eller ventrikkeltakykardi (VT) med venstre grenblokk-morfologi er blant de vanligste kliniske manifestasjonene. ARVC er sjelden hos barn, men de yngste ARVC-pasientene har ofte en mer alvorlig fenotype med høy risiko for plutselig hjertedød og progresjon mot hjertesvikt (13). Dessverre kan plutselig død være det første symptomet på ARVC, og sykdommen er den vanligste årsaken til plutselig død blant unge i Skandinavia (14).
Behandling og oppfølging
Behandlingen av ARVC-pasienter har som mål å forhindre plutselig hjertedød og å redusere eventuelle arytmi- og hjertesviktsymptomer. Betablokker skal vurderes hos alle pasienter med ARVC, og det anbefales å gi høyeste tolerable terapeutiske dose. Dette supprimerer ventrikulær arytmi, men det er ikke vist at medikamentell behandling gir bedret overlevelse. Hos barn er vanlig betablokker (metoprolol) førstevalg, deretter sotalol og ev. kombinasjon av metoprolol og flekainid. Ved bruk av flekainid må man være oppmerksom på ev. vedvarende lavfrekvent VT. Amiodaron brukes lite pga. langtidsbivirkninger. Ved hjertesvikt gis i tillegg diuretika og angiotensinkonverterende enzym (ACE)-hemmer.
Forebygging av plutselig hjertedød
ICD-indikasjon hos barn er i utgangspunktet gjennomgått hjertestans og livstruende vedvarende VT, samt primærprofylaktisk i påvente av en hjertetransplantasjon ved alvorlig hjertesvikt. Det er utviklet en risikokalkulator, men denne er ikke validert for personer < 16 år (15).
Konsekvenser for idrett
Vi har lite kunnskap om hvilken betydning fysisk aktivitet har for sykdomsutviklingen hos barn med ARVC, og det gis i utgangspunktet ingen restriksjoner på fysisk aktivitet så lenge det ikke er tegn til sykdomsmanifestasjon. En nederlandsk studie viste imidlertid at utvikling av ARVC i barne- og ungdomsalder var assosiert med deltagelse i utholdenhetsidrett og at pasienter som utviklet ARVC i ungdomsårene, hadde flere årlige timer med trening i barnealder sammenlignet med pasienter som utviklet ARVC i voksen alder (16). Studier på voksne ARVC-pasienter har vist at høyintensitetsidrett er forbundet med både tidligere arytmidebut og mer alvorlig sykdomsforløp (17). Høyintensitetsidretter anbefales derfor ikke hos barn med genvarianter forbundet med ARVC.
Familiescreening
I retningslinjene fra ESC anbefales det å vurdere screening av asymptomatiske familiemedlemmer, inkludert barn, på det tidspunktet ARVC er diagnostisert i familien. En studie fra Barnekardiologisk avdeling på Rikshospitalet viste en sykdomspenetrans på 18 % hos genotype-positive barn som gjennomgikk familiescreening (13). Genotypepositive slektninger bør følges opp med kardiologiske kontroller årlig eller hvert 2. år, inkludert EKG, ekkokardiografi og Holter-registrering. MR av hjertet har også en fremtredende rolle i evalueringen (2).
Restriktiv kardiomyopati
Restriktiv kardiomyopati (RCM) er en sjelden form for myokardsykdom karakterisert ved unormal diastolisk funksjon med restriktiv fysiologi og normal ventrikkeldiameter, veggtykkelse og systolisk funksjon (1). Mens to tredeler av barna har ren RCM-fenotype, har den resterende delen en blandet RCM-HCM-fenotype (18).
RCM er den mest sjeldne formen for kardiomyoapti hos barn med en insidens på 0,03–0,04 per 100 000 barn (2). Alder ved sykdomsdebut varierer fra tidlig spedbarnsalder til sen voksen alder.
Diagnostikk og utredning
RCM kan være arvelig, idiopatisk eller knyttet til andre sykdommer som f.eks. amyloidose eller Friedreich ataksi, men kan også være legemiddelindusert eller skyldes stråling. Ekkokardiografi er det viktigste diagnostiske verktøyet og viser typisk bilateralt betydelig dilaterte atrier og uttalt diastolisk dysfunksjon med forhøyede fylningstrykk. Hjertekateterisering bør utføres der det er tvil om diagnosen. Hjerte-MR skiller RCM fra konstruktiv perikarditt og gir informasjon om det foreligger myokardfibrose.
Utredningen bør også innebefatte leting etter ekstrakardiale manifestasjoner, inkludert symptomer på nevromuskulær sykdom.
Genetikk
Når den er arvelig, presenterer RCM seg oftest som en autosomal dominant lidelse. Gener assosiert med RCM koder ofte for hjertesarkomerproteiner. Selv om alle store sarkomeriske gener kan forårsake RCM, er det vanligste sykdomsgenet TNNI3. Mutasjoner i desmin-genet kan også gi restriktiv kardiomyopati i kombinasjon muskeldystrofi.
Symptomer og funn
Tidlige symptomer på RCM kan være uspesifikke, inkludert generell tretthet og redusert fysisk yteevne. Kliniske funn sekundært til forhøyet systemisk og pulmonalt venetrykk kan inkludere tydelig halsvenepuls, perifert ødem, hepatomegali, lungeødem og pulmonal hypertensjon. Forhøyet pulmonal vaskulær motstand foreligger hos opptil 40 % av barn med RCM og kan stige raskt selv i fravær av andre kliniske endringer, noe som har betydning for timing av hjertetransplantasjon. Synkope er et uspesifikt, men illevarslende debutsymptom som kan skyldes arytmier eller tromboemboliske hendelser. Barn med RCM har dårligst prognose av alle barn med kardiomyopati. 78 % av barn med ren RCM gjennomgår hjertetransplantasjon innen 5 år etter at de fikk diagnosen (18).
Behandling og oppfølging
Antikoagulasjon anbefales pga. risiko for tromboemboliske komplikasjoner, men forøvrig er behandlingen kun symptomatisk. Det anbefales å starte utredning med tanke på hjertetransplantasjon på et forholdsvis tidlig stadium.
Familiescreening
I henhold til de nye ESC-retningslinjene anbefales det at barn gentestes på lik linje med andre familiemedlemmer, uavhengig av alder, og at oppfølgingen individualiseres i forhold til ev. forekomst av tidlig sykdomsdebut i familien og forventet utvikling av sykdommen (2). Det er viktig å være klar over at RCM kan forekomme hos personer med en familiehistorie med HCM.
Oppfølging ved senter med spesialkompetanse versus lokal oppfølging
Diagnostikk, behandling og oppfølging av genetiske hjertesykdommer er som hovedregel en kardiologisk spesialistoppgave og bør med fordel utføres av et dedikert team. De barnekardiologiske miljøene rundt om i landet er små, med varierende erfaring og kunnskap. I noen geografiske områder er det økt prevalens av enkelte genetiske sykdommer. Dette krever økt lokal ekspertise.
Barnekardiologisk avdeling ved Oslo universitetssykehus, Rikshospitalet ønsker at barn med genotyper assosiert med høy risiko for plutselig hjertedød skal henvises for sentral oppfølging. Det er snakk om et fåtalls pasienter som er spredt rundt om i landet, og hvor det å kunne støtte seg på et team av genetikere, elektrofysiologer og radiologer er et stort fortrinn som behandler. Barnekardiologisk avdeling samarbeider også tett med fagmiljøet tilknyttet Kardiogenetisk poliklinikk ved Kardiologisk avdeling på OUS, Rikshospitalet, både klinisk, med regelmessige møter hvor pasienter diskuteres i et multidisiplinært forum, og gjennom felles forskningsprosjekter.
Referanser og litteratur
- Lipshultz SE, et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation 2019; 140:e9-e68.
- Arbelo E, et al. 2023 ESC Guidelines for the management of cardiomyopathies. European heart journal 2023; 44:3503-626.
- Marston NA, et al. Clinical characteristics and outcomes in childhood-onset hypertrophic cardiomyopathy. European heart journal 2021; 42:1988-96.
- Lafreniere-Roula M, et al. Family screening for hypertrophic cardiomyopathy: Is it time to change practice guidelines? European heart journal 2019; 40:3672-81.
- Page SP, et al. Cardiac myosin binding protein-C mutations in families with hypertrophic cardiomyopathy: disease expression in relation to age, gender, and long term outcome. Circ Cardiovasc Genet 2012; 5:156-66.
- Norrish G, et al. Yield of Clinical Screening for Hypertrophic Cardiomyopathy in Child First-Degree Relatives. Circulation 2019; 140:184-92.
- Cardim N, et al. Role of multimodality cardiac imaging in the management of patients with hypertrophic cardiomyopathy: an expert consensus of the European Association of Cardiovascular Imaging Endorsed by the Saudi Heart Association. Eur Heart J Cardiovasc Imaging 2015; 16:280.
- Norrish G, et al. Development of a Novel Risk Prediction Model for Sudden Cardiac Death in Childhood Hypertrophic Cardiomyopathy (HCM Risk-Kids). JAMA Cardiol 2019; 4:918-27.
- Forså MI, et al. Distinguishing left ventricular hypertrophy from hypertrophic cardiomyopathy in adolescents: a longitudinal observation study. Eur J Prev Cardiol 2024; 31:591-8.
- Belkaya S, et al. Autosomal Recessive Cardiomyopathy Presenting as Acute Myocarditis. Journal of the American College of Cardiology 2017; 69:1653-65.
- Skjølsvik ET, et al. Exercise is Associated With Impaired Left Ventricular Systolic Function in Patients With Lamin A/C Genotype. Journal of the American Heart Association 2020; 9:e012937.
- Marcus FI, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation 2010; 121:1533-41.
- Smedsrud MK, et al. Highly malignant disease in childhood-onset arrhythmogenic right ventricular cardiomyopathy. European heart journal 2022.
- Holst AG, et al. Incidence and etiology of sports-related sudden cardiac death in Denmark--implications for preparticipation screening. Heart rhythm 2010; 7:1365-71.
- Cadrin-Tourigny J, et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. European heart journal 2022.
- Te Riele A, et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy in the Pediatric Population: Clinical Characterization and Comparison With Adult-Onset Disease. JACC Clinical electrophysiology 2015; 1:551-60.
- Lie Ø H, et al. Harmful Effects of Exercise Intensity and Exercise Duration in Patients With Arrhythmogenic Cardiomyopathy. JACC Clinical electrophysiology 2018; 4:744-53.
- Webber SA, et al. Outcomes of restrictive cardiomyopathy in childhood and the influence of phenotype: a report from the Pediatric Cardiomyopathy Registry. Circulation 2012; 126:1237-44.
Andre norske dokumenter å kunne støtte seg på
Tidligere versjoner
2018: Jakob Klcovansky og Thomas Möller