









Generell veileder i pediatri
8. Hjerte- og karsykdommer
8.16 Pulmonal hypertensjon
Sist faglig oppdatert: 27.07.2022
Henrik Brun og Gottfried Greve
Bakgrunn
Idiopatisk pulmonal arteriell hypertensjon (IPAH) er svært sjelden i barnealder med antatt insidens på 1 per million per år og en prevalens på ca 4 per million, med ca. dobbelt så høy forekomst hos kvinner. Debut er vanligst i ung voksen alder.
Sekundær pulmonal arteriell hypertensjon (PAH) er vanligste årsak til pulmonal hypertensjon (PH) i barneårene. Forekomsten av PAH sekundært til hjertefeil avhenger av tilgjengelighet og kvalitet på barnekardiologisk og kirurgisk helsetjeneste. Ca. 5 % av alle voksne med medfødte hjertefeil utvikler PAH. Prevalens av PAH assosiert med medfødt hjertefeil er estimert til mellom 1,6 til 12,5 per million, hvorav 25–50 % har Eisenmengers syndrom. Ikke opererte medfødte hjertefeil fører til PAH i varierende frekvens og tempo. Transposisjon med stor VSD og truncus arteriosus kompliseres oftest med PAH, fulgt av non-restriktiv VSD.
I Norge ble det i perioden 1990–2002 registrert drøyt 100 barn med PAH. Ca 20 % av disse hadde Down syndrom.
Pulmonal hypertensjon relatert til lungesykdom utgjør også en signifikant andel og har fått økende oppmerksomhet hos barn de siste årene. Blant annet kan prostasyklinbehandling ha en rolle ved PH relatert til bronkopulmonal dysplasi (BPD) hos prematurt fødte barn.
Definisjon og inndeling
Pulmonal hypertensjon defineres, basert på kardiologisk tradisjon, som et middeltrykk i lungearterien på over 25 mmHg i hvile. WSPH (World Symposium in PH) inndeling av PAH (oppdatert sist 2013) er kun delvis egnet for barn. En egen, detaljert inndeling for barn ble foreslått i 2011 (Panama-klassifikasjonen), men er under revisjon og ikke i aktiv bruk.
| Hovedgruppe | Undergrupper | Relevans for barn |
| 1. Pulmonal arteriell hypertensjon (PAH) | 1.1 Idiopatisk 1.2 Arvelig 1.2.1 BMPR-2-mutasjon 1.2.2 ALK1, endoglin-mutasjon (HHT) 1.2.3 Ukjent gen 1.3 Medikament/toksin-indusert 1.4 Assosiert med: 1.4.1 Bindevevssykdom 1.4.2 HIV infeksjon 1.4.3 Portal hypertensjon 1.4.4 Medfødt hjertefeil 1.4.5 Schistosomiasis 1.4.6 Kronisk hemolytisk anemi 1.5 Persisterende pulmonal hypertensjon hos nyfødte 1.6 Pulmonal veno-okklusiv sykdom og/eller pulmonal kapillær hemangiomatose (assosiert med mutasjon i EIF2AK4 gen) | 40–50 % av PAH hos barn er assosiert med medfødt hjertefeil. I vår del av verden utgjør idiopatisk og arvelig PAH den andre halvparten. PAH ved HIV, ved portal hypertensjon og relatert til bindevevssykdommer forekommer hos barn, men er sjelden |
| 2. Pulmonal hypertensjon relatert til venstresidig hjertesykdom | 2.1 Systolisk dysfunksjon 2.2 Diastolisk dysfunksjon 2.3 Klaffesykdom | F.eks. mitralstenose og lungevenestenose samt venstre ventrikkel dysfunksjon |
| 3. Pulmonal hypertensjon relatert til lungesykdom og eller hypoksemi | 3.1 Kronisk lungesykdom 3.2 Interstitiell lungesykdom 3.3 Annen lungesykdom med blandet obstruktivt/restriktivt mønster 3.4 Søvnapne 3.5 Alveolær hypoventilasjon 3.6 Langvarig høydeopphold 3.7 Utviklingsforstyrrelser lunge | F.eks. diafragmahernie BPD Øvre luftveisobstruksjon Lungehypoplasi (Begge de siste relevante ved Downs syndrom) |
| 4. Kronisk tromboembolisk pulmonal hypertensjon | Ekstremt sjelden hos barn | |
| 5. Pulmonal hypertensjon med uklar multifaktoriell genese | 5.1 Hematologiske tilstander 5.2 Systemsykdommer: sarkoidose, pulmonal Langerhans celle histiocytose, Lymfangioleiomyomatose, nevrofibromatose, vaskulitt 5.3 Metabolsk sykdom: glykogenose, Gaucher, Tyreoideasykdom 5.4 andre: Tumorobstruksjon, fibroserende mediastinitt, kronisk nyresvikt/dialyse |
Symptomer og funn
Vag klinikk, med anstrengelsesdyspnø som ofte forveksles med astma i startfasen. Barn har oftere synkope og sjeldnere hjertesvikt med ødem ved diagnose. Hos helt små barn sees redusert trivsel/vekst og aktivitetsnivå. Cyanose sees ved kardial eller intrapulmonal shunt og noen ganger kun ved anstrengelser. Det er vanlig med “doctor's delay”.
Diagnostikk og utredning
Undersøkelser
Ekkokardiografi er egnet for screening og oppfølging av PAH:
- Systolisk trykk i høyre ventrikkel estimeres fra maksimal Doppler-hastighet i tricuspidalinsuffisiensen pluss det systoliske trykk i høyre atrium. Under forutsetning av at det ikke er pulmonalstenose vil systolisk trykk i høyre ventrikkel være lik systolisk trykk i lungearterien.
- Middeltrykk i lungearterien kan estimeres ut fra maksimal estimert trykkgradient i pulmonalinsuffisiensen pluss høyre atrietrykk (vanligvis 5 mmHg)
- Ved shunt i høytrykksområdet vil shuntens retning og maksimale systolisk hastighet uttrykke forskjell i systolisk blodtrykk mellom de aktuelle deler av pulmonal- og systemisk kretsløp. Maksimal hastighet og retning av blodstrømmen i åpenstående ductus arteriosus gir for eksempel et direkte mål på trykkforskjell mellom aorta og pulmonalarterien og PH kan diagnostiseres non-invasivt.
- Blodstrøm i en ASD forteller ikke direkte om trykk i lungekretsløpet, men om compliance og fylningstrykk av atrier og ventrikler
Eisenmenger-diagnose er oftest lett å stille non-invasivt med ekkokardiografi på basis av non-restriktiv shunt og cyanose. For de fleste andre problemstillinger vil det være aktuelt å estimere lungekarmotstand eller forhold mellom lungekarmotstand og systemisk karmotstand ved høyre hjertekateterisering. Lungekarmotstand må sees i forhold til høyre ventrikkels pumpeevne.
For pasienter med en funksjonell ventrikkel (Fontanopererte) vil kun en liten økning i lungekarmotstanden ha store konsekvenser i form av redusert minuttvolum. Dette er vanskelig å diagnostisere ekkokardiografisk og krever invasive mål.
Høyresidig hjertekateterisering brukes for å estimere lungekarmotstanden og dens reversibilitet:
- Minuttvolum gjennom lungene kalkuleres ved Fick’s prinsipp eller hvis det ikke er shunting av blod, ved termodilusjon. Ved PDA må denne ballongokkluderes.
- Lungekarmotstanden kalkuleres ved differansen mellom middeltrykket i venstre atrium (kiletrykk) og middeltrykk i lungearterien delt på hjertets minuttvolum gjennom lungene.
- Vasodilatasjonstest gjøres med 100 % oksygen, med NO på intubert pasient eller med prostanoider. Respons er definert som mer enn 20 % reduksjon i lungekarmotstand eller lungearterietrykk. Vasodilatorrespons kan oppstå etter lengre tid med medikamentell behandling. Ved idiopatisk/arvelig PAH og (nær) normalisering av lungekarmotstanden under testing (sjelden) skal en starte behandling med kalsiumkanalblokker. Disse kan imidlertid senere miste effekt.
- Risiko ved hjertekateterisering øker med bruk av narkose, med økende lungekarmotstand og dårlig funksjonsklasse (WHO IV). Resultater ved sentre med god erfaring og anestesiservice viser imidlertid at risiko for alvorlige komplikasjoner er lav (1 %).
Belastning på tredemølle med måling av O2-opptak. Resultater av en slik undersøkelse korrelerer med trykk i høyre atrium og lungearterien og gir prognostisk informasjon. Manglende dilatasjon av lungekar ved anstrengelse gir redusert hjerteminuttvolum og maksimalt O2-opptak. Betydelig fall i O2-metning sees ved åpne shunter fordi vasodilatasjon i aktiv skjelettmuskel gir fall i systemisk karmotstand og derved økende høyre til venstre shunt. Dette sees ved Eisenmenger syndrom. Ved Eisenmenger syndrom vil perifer måling av O2-metning være en indikator for endringer i forholdet mellom lungekarmotstanden og systemisk karmotstand. Ved åpen ductus arteriosus må målinger gjøres postductalt. Resultater påvirkes også av endret minuttvolum via oksygenmetning i venøst returblod.
Fig. 1: Diagnostisk algoritme fra Europeiske guidelines 2019
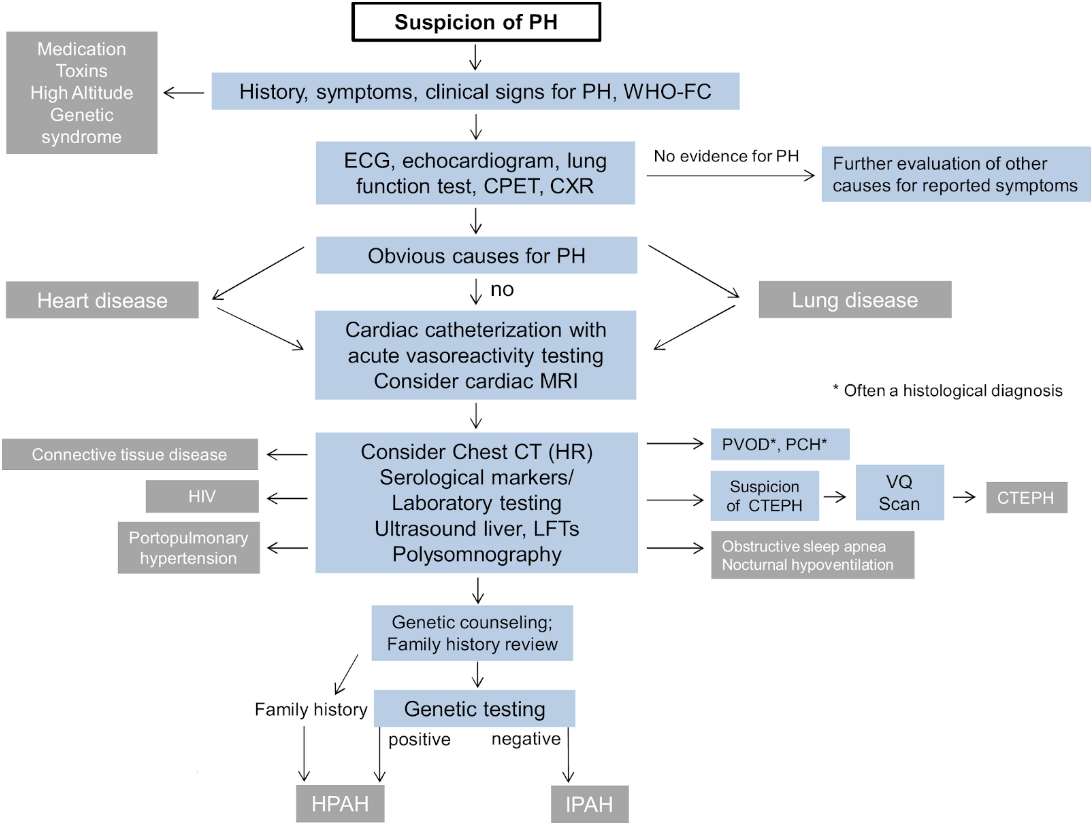
Behandling og oppfølging
I 2015 kom AHA og ATS med de første pediatriske guidelines for PH utredning og behandling. Europeisk versjon fra European Pediatric PVD network (PPVDN) kom i 2016, oppdatert i 2019 (referanse 3). Herfra er hentet tabeller for diagnose, risikostratifisering og behandlingsalgoritme. Egen algoritme for BPD relatert sykdom er publisert.
Fig 2 Behandlingsalgoritme, PH hos barn (PPVDN 2019)
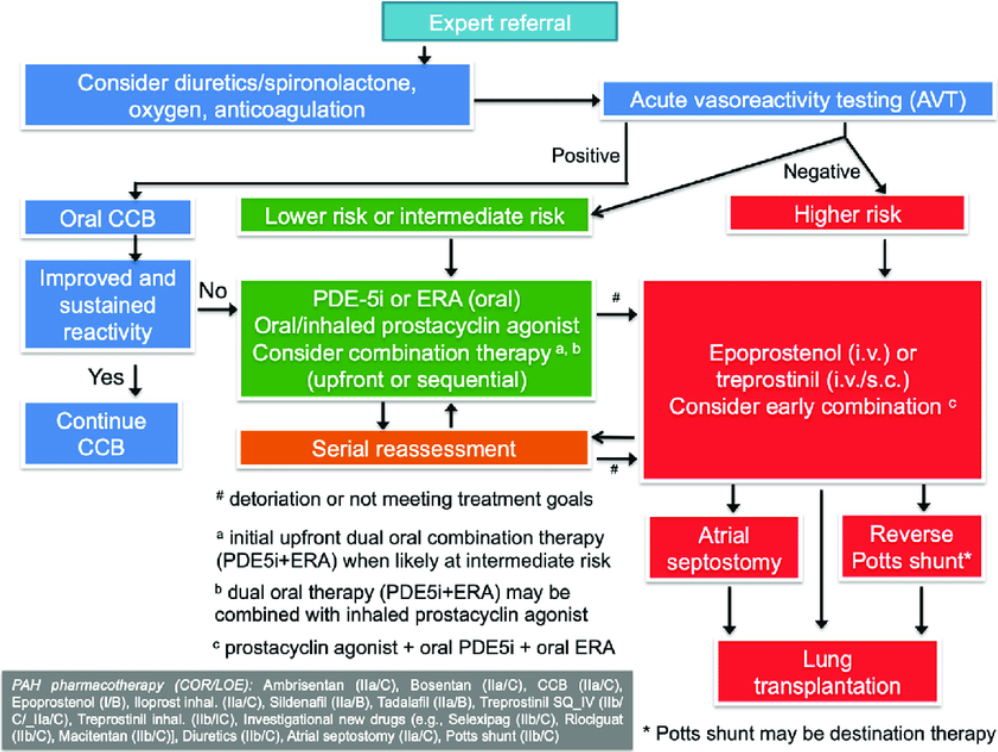
Europeiske guidelines (ESC) anbefaler at guidelines for voksne tas i betraktning også ved behandling av barn (anbefaling klasse IIa)
Såkalt avansert behandling krever spesialkompetanse, spesielt ved oppstart av behandlingen. Forebyggende/støttende tiltak og sikkerhetsregler bør kjennes av alt helsepersonell som møter disse pasientene.
Støttende og forebyggende behandling
Generelt bør alle pasientene unngå:
- Hard eller ekstrem isometrisk fysisk aktivitet
- Dehydrering
- Graviditet
- Opphold i store høyder. Flyturer i trykkabin tolereres av mange, men noen trenger ekstra oksygen. Det kan evt. undersøkes på forhånd med en såkalt HAST-test der pasienten puster inn en gassblanding med redusert O2 innhold. Ta evt. kontakt med Flymedisinsk institutt i Oslo.
- Luftveissykdommer. Det anbefales derfor influensa- og pneumokokkvaksine til alle og RS-virus immunglobulin (palivizumab) til de minste. Lav terskel for innleggelse og oksygentilførsel.
Kirurgi og anestesi bør så langt som overhodet mulig finne sted ved OUS-Rikshospitalet.
Oksygenbehandling
Nattlig eller kontinuerlig hjemmeoksygenbehandling kan redusere lungekarmotstand og muligens øke overlevelse og forsinke polycytemiutvikling ved Eisenmenger syndrom, men data er usikre og behandlingen har store praktiske konsekvenser. Ved høyre hjertesvikt med økt oksygenekstraksjon i hvile og ved akutte luftveisinfeksjoner er oksygen også til nytte. En vanlig brukt cut-off for SpO2 er 92 % men lavere kan aksepteres bla ved BPD. Vi tester ut og legger vekt på symptomlindring for tretthet, morgenhodepine, energinivå og andre symptom på lavt oksygentilførsel til vev og organer
Hyperviskositetssyndrom (gjelder bare Eisenmenger)
Høy hematokrit er en følge av lav oksygenmetning og vil i seg selv øke lungekarmotstand. Venesectio bidrar til å redusere hematokrit, men øker i seg selv risikoen for slag. Venesectio er derfor kun aktuelt ved alvorlig symptomgivende hyperviskositetssyndrom og hematokrit over 70 %. Tappet blod erstattes med krystalloid. Det er også aktuelt før kirurgi etter hematologisk vurdering med tromboelastografi. Jernbehandling er indisert hos pasienter med jernmangel på tross at de har høy hematokrit. Vi følger skjema for forventet Hb/Hct i relasjon til SpO2 i hvile.
Sviktbehandling
En bør være varsom med intensiv diuretikabehandling fordi pasientene er avhengige av intravasal fylning for å opprettholde hjertets minuttvolum. Dehydrering øker komplikasjonsfarene ved erytrocytose og hyperviskositet. Både diuretika og digitalisering er aktuelt ved høyre hjertesvikt. Følg NTproBNP.
Antikoagulasjon
Warfarin er alltid indisert ved kronisk tromboembolisk PH og kan være indisert ved idiopatisk/arvelig PAH. Marevanbehandling er forbundet med mer komplikasjoner enn nytte ved Eisenmenger syndrom, der pasienter samtidig har økt blødnings- og tromboserisiko. En del pasienter med Eisenmenger får ASA, men effekten er dårlig dokumentert.
Videre avansert medikamentell behandling
Kalsiumkanalblokkere
Nifedipin er kun aktuelt ved normalisering av lungekarmotstanden ved reversibilitetstesting (sjelden) hos IPAH pasienter, og anvendes i praksis ikke til barn.
Prostasykliner og PG agonister
Langtidsbehandling med treprostinil (Remodulin®) intravenøst eller subkutant har den mest potente vasodilaterende effekten ved PAH, også hos pasienter uten vasodilatorrespons under hjertekateterisering. Medikamentet har middels kort halveringstid og krever kontinuerlig infusjon. Medikamentet er førstevalg ved alvorlig PAH (funksjonsklasse IV). Treprostinil kan gis subkutant via implantert pumpe med månedlig transkutan fylling (fra ca. 30 kg kroppsvekt) eller som kontinuerlig subkutan infusjon med feks I-jet pumpe tilsvarende insulinpumpe.
Inhalasjon av iloprost (Ilomedin®) har tilsvarende effekt ved PAH. Kort halveringstid krever 6–12 inhalasjoner per dag. Dette gjør langtidsbehandling lite aktuell for barn. Selexipag (Uptravi®) er en ny non-prostanoid peroral PGI2 agonist under med epoprostenol-lik effekt, godkjent for voksne i WHO klasse II-III også med PAH relatert til medfødte hjertefeil. En liten pediatrisk studie med snittalder 7 år antyder akseptabel effekt og sikkerhet.
Endotelinreseptorblokkere
Bosentan (Tracleer®), kompetitiv endotelin reseptor A og B antagonist, gis peroralt to ganger daglig. Sitaxentan er en mer selektiv reseptor A antagonist og ambrisentan enda mer selektiv. De to siste er lite prøvet på barn så langt. Macitentan (Opsumit®) (A+B) i samme gruppe skal være mer vevs-selektiv. En liten studie 0–18 år antyder akseptabel sikkerhet og god effekt.
Bosentan har vist klinisk og hemodynamisk effekt i to kontrollerte studier ved Eisenmenger syndrom og er førstevalg ved denne tilstanden. Retrospektive studier støtter bedret langtidsoverlevelse ved avansert medikamentell behandling av voksne med Eisenmenger-syndrom, og flere sentra starter nå behandling av pasienter i WHO funksjonsklasse II.
Fosfodiesterasehemmere
Sildenafil (Revatio®) gis peroralt 3 ganger daglig. Standard voksen dose er 20 mg x 3. Sildenafil har additiv effekt gitt sammen med bosentan og prostanoider, men interaksjon med bosentan reduserer effekt noe. Høy dose var assosiert med økt dødelighet i STARTS studien. Tadalafil har lengre halveringstid og mindre interaksjonsproblemer. En liten studie med pasienter 4–18 år antyder akseptabel sikkerhet og god effekt av tadalafil.
Kombinasjonsbehandling
Medikamentgruppene i) prostanoider, ii) endotelinreseptorblokkere og iii) fosfodiesterasehemmere påvirker patofysiologien ulikt. Ulike kombinasjoner av behandling er rapportert for barn og dokumentert effektivt og trygt hos voksne. Medikamentgruppene kan kombineres i duo- og ved ”ryggen mot veggen” trippelbehandling. Interaksjoner og bivirkninger må da påregnes, særlig hvis pasienten warfarin-behandles i tillegg.
Spesialistoppgave å vurdere slik behandling. Interaksjonsproblemene er størst for kombinasjonen bosentan og sildenafil som er den vanligste brukte. Bosentan reduserer effekten av sildenafil som da må doseres høyere. Mindre interaksjon med tadalafil og macitentan samt dosering x 1 per dag gjør at denne kombinasjonen er økende brukt også i barnepopulasjonen. Kombinasjonsbehandling skal vurderes fra WSPH klasse II-III ved diagnose.
Kirurgisk og intervensjonell behandling
- Intervensjonell atrieseptostomi er indisert ved synkope. Redusert oksygenmetning p.g.a. høyre til venstre shunt tåles pga bedret fylning av venstre ventrikkel og dermed økt minuttvolum. Behandlingen er aktuell ved alvorlig PAH som ikke svarer tilstrekkelig på Remodulin. Risiko ved inngrepet reduseres ved ballonggradert shunt. På barn har vi brukt såkalt diabolostent som kan tilpasses i størrelse. Alle med høyre-venstre atrieshunt skal ha ASA profylakse.
- Anleggelse av en Pott’s shunt (tilsvarer en stor ductus) er et alternativ til atrieseptostomi som er under utprøving. Fordelen er preservert O2-metning til hjernen.
- Lungetransplantasjon eller hjerte- og lungetransplantasjon er siste valg.
- Pulmonal trombendarterektomi er sjelden aktuelt hos barn.
- Lungetransplantasjon gjøres, men er sjelden et behandlingsvalg hos barn. Men tilbudet er aktuelt hos ungdom og unge voksne.
Behandlingsmål
Behandlingsrespons er vanskelig å evaluere.
Hos voksne og større barn anbefales målstyrt behandling med predefinerte, sammensatte behandlingsmål.
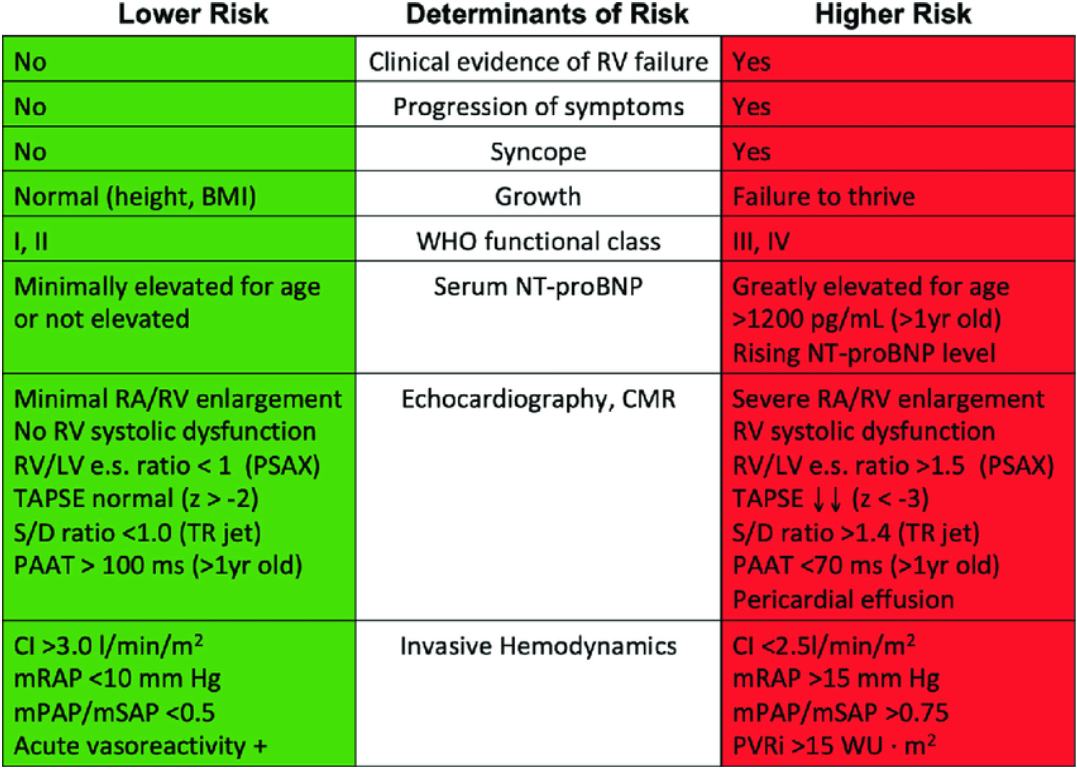
Hos mindre barn styres behandlingen etter klinisk evaluering, inkludert standardisert oksygenmetningsmålinger, tester av fysisk yteevne (tredemølle og 6 minutters gangtest), proBNP-verdier og ekkokardiografiske mål (trikuspidalinsuffisiens maksimalhastighet, TAPSE (Tricuspid annular plane systolic excursion), høyre atriums størrelse, høyre ventrikkel arealmål). Behandlingsmålene reflekteres i tabell under for risikostratifisering og behandlingsstyring der mål er å beholde pasienten i lavrisiko.
Fig 3 Risikogruppering PH hos barn (PPVDN 2019)
Prognose
Forventet levetid ved ubehandlet IPAH er muligens kortere for barn enn for voksne. Rapportert naturlig forløp er 1–2 år forventet levetid etter symptomdebut. Vesentlig forlenget levetid er dokumentert ved avansert medikamentell behandling. Ved PAH sekundært til hjertefeil er overlevelse bedre, dog med kortere forventet levetid ved komplekse hjertefeil enn ved enkle. I tillegg er ofte livskvaliteten ved Eisenmenger syndrom betydelig redusert med stor komorbiditet knyttet til cyanose/erytrocytose og tromboemboliske komplikasjoner. Hos enkelte barn sees rask utvikling av PAH tross en hemodynamisk lite belastende hjertefeil, f.eks. ASD. Den genetiske bakgrunn er her sannsynligvis avgjørende, og tilstanden behandles som primær PAH. Hos andre sees utvikling av PAH etter kirurgisk korreksjon av hjertefeil. Disse pasientene har også utbytte av avansert medikamentell behandling.
Referanser
- Dimopoulos K et al. Improved Survival among patients with Eisenmenger Syndrome receiving advanced therapy for pulmonary arterial hypertension. Circulation 2010; 121:20-25
- Galie N et al. The use of combination therapy in pulmonary arterial hypertension: new developments. Eur Respir Rev 2009; 18: 113, 148-153
- Hansmann G. et al. 2019 updated consensus statement on the diagnosis and treatment of pediatric pulmonary hypertension: The European Pediatric Pulmonary Vascular Disease Network (EPPVDN), endorsed by AEPC, ESPR and ISHLT. J Heart Lung Transplant 2019;38: 879-901
- Hansmann G. et al. Selexipag for the treatment of children with pulmonary arterial hypertension: First multicenter experience in drug safety and efficacy. J Heart Lung Transplant 2020; 39:695-706
- Schweintzger S et al. Safety and efficacy of the endothelin receptor antagonist macitentan in pediatric pulmonary hypertension. Cardiovasc Diagn Ther. 2020; 10: 1675-85
- Takatsuki S et al. Initial experience with Tadalafil in Pediatric Pulmonary Arterial Hypertension. Pediatr Cardiol. 2012; 33: 683–8.
- Hansmann G et al. Pulmonary hypertension in bronchopulmonary dysplasia. Pediatr Res 2020 doi: 10.1038/s41390-020-0993-4. Online ahead of print
Ressurser
- Pediatric Pulmonary Vascular Disease Network: Europeisk non profit interesseorganisasjon med oppdatert nettside. https://www.pvdnetwork.org/
- Uptodate: https://www.uptodate.com/contents/pulmonary-hypertension-in-children-classification-evaluation-and-diagnosis
Tidligere versjoner:
Publisert 2006: Gottfried Greve, Henrik Brun og Henrik Holmstrøm
Revidert 2011: Henrik Brun, Gottfried Greve og Henrik Holmstrøm