









Nyfødtveileder
8 Gulsott og hemolytisk sykdom
8.4 Langvarig gulsott hos nyfødte og spedbarn
Sist faglig oppdatert: 24.04.2026
Claus Klingenberg, Astri Lang, Ketil Størdal, Anlaug Vatne, Asborg Aanstad Bjertnæs, Eline Vold Engesli, Erling Tjora, Ragnhild Støen, Anniken Bjørnstad Østensen og Runar Almaas
Bakgrunn
- Langvarig gulsott defineres som synlig gulsott
- Ved 2 ukers alder hos barn som kun får morsmelkerstatning
- Ved 3 ukers alder hos barn som ammes + ev. noe tillegg
- Langvarig gulsott deles inn i:
- Ukonjugert type: Vanligst, og oftest morsmelk-assosiert gulsott
- Konjugert type: Sjeldnere, men kan indikere intra- eller ekstrahepatisk gallestase som er viktig å fange opp tidlig
Langvarig gulsott av UKONJUGERT type
- Morsmelk-assosiert gulsott er aller vanligst.
- Andre årsaker: Hemolyse pga. immunisering, hypotyreose og medfødt infeksjon (CMV)
- Erytrocytt-cellemembrandefekter (hereditær sfærocytose, elliptocytose) og hemoglobinopatier debuterer sjelden i nyfødtperioden.
- Kan en sjelden gang også skyldes tilstander som Gilbert syndrom, G6PD-mangel, pyruvat kinase mangel, galaktosemi, Crigler-Najjar syndrom, tyrosinemi.
Nærmere om «morsmelk-assosiert gulsott»
- Dette er en gulsott av ukonjugert type, antagelig multifaktoriell, og ufarlig.
- Rapportert hos inntil 15–30 % av barn som fullammes ved 3–4 ukers alder.
- Vanligvis økende verdier av total (ukonjugert) bilirubin fra 4.–7. levedøgn.
- Kan medføre høye total bilirubin-verdier i 2. og 3. leveuke.
- Barna er ellers friske, ev. litt søvnige, og har normal farget avføring og lys urin.
- Morsmelk-assosiert gulsott kan vedvare i 3–12 uker. Lysbehandles ved behov etter vanlige retningslinjer.
- Å bytte ut morsmelk med morsmelkstillegg er ikke lenger anbefalt.
- Etter 3–4 ukers alder er morsmelk-assosiert gulsott en eksklusjonsdiagnose, og man skal måle total og direkte (konjugert) bilirubin (se under).
Langvarig gulsott av KONJUGERT type
- Viktig å utelukke ekstrahepatisk gallegangsatresi (og koledokuscyste)
- Andre årsaker (rekkefølge etter antatt hyppighet): Langvarig parenteral ernæring, medfødte infeksjoner (inkl. CMV), alfa-1-antitrypsinmangel, kolestase pga. koledokuscyste eller ”inspissated bile» (oftest etter hemolyse), galaktosemi (gir vanligvis mest ukonjugert hyperbilirubinemi), cystisk fibrose (kan gi steatose i spedbarnsalder, men oppdages ofte på nyfødtscreening), Aagenæs syndrom, metabolsk sykdom inkl. defekter i syntese av gallesyrer, mitokondriesykdommer, kromosomfeil (trisomi 13, 18, 21 osv.), hypofysesvikt, progressiv familiær intrahepatisk kolestase, Alagille syndrom, Zellwegers sykdom (cerebrohepatorenalt syndrom), GALD m.fl.
Symptomer og funn
I de første ukene er de fleste pasienter med kolestase nesten asymptomatiske og det kan ta tid før man ser dårlig vektutvikling.
Viktig å vurdere:
- Farge på avføring og urin.
- Hvit/kittaktig avføring og mørk urin tyder på manglende utskillelse av konjugert bilirubin fra lever til tarm og bør utløse rask henvisning til barneavdeling.
- Type ernæring (morsmelk vs. erstatning)
- Vektutvikling
- Fødselsvekt (SGA)
- Langvarig parenteral ernæring
- Lever og milt (hepatosplenomegali)
- Hjerte bilyd (pulmonalstenose/Fallot ved Alagille syndrom)
- Andre stigmata/anomalier
- Utslett/hudblødninger (medfødt CMV)
- Familieanamnese på blodsykdom eller neonatal ikterus
Diagnostikk og utredning
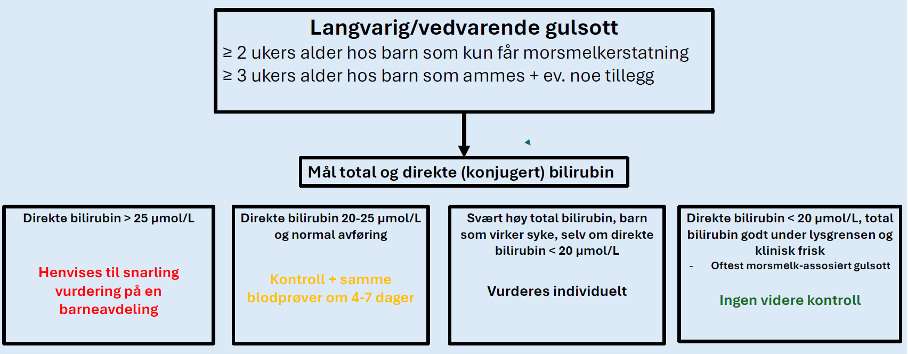
Vi anbefaler at det måles total og direkte (konjugert) bilirubin hvis det er synlig gulsott:
- Ved 2 ukers alder hos barn som kun får morsmelkerstatning
- Ved 3 ukers alder hos barn som ammes + ev. noe tillegg
Hvis barnet ellers virker frisk, anbefaler vi ikke å ta andre blodprøver.
NB. På de fleste laboratorier besvares «direkte bilirubin» som «konjugert bilirubin». Imidlertid måles, på alle laboratorier i Norge, direkte bilirubin med en metode som gjør at direkte bilirubin blir høyere hvis også total bilirubin er høy. Det medfører at hvis man f.eks. ved en morsmelk-assosiert gulsott har høye total bilirubin verdier, vil relativt sett også direkte bilirubin bli høyere uten at det er uttrykk for kolestase. En bør derfor alltid også se på total bilirubin verdien når man vurderer konjugert bilirubin. I noen land anbefales å bruke «prosentandel direkte/total bilirubin», og noen har hatt < 20 % som liten mistanke for kolestase. Andre land og sentrale retningslinjer benytter ikke en slik prosentandel.
Følgende barn skal henvises til snarlig vurdering på en barneavdeling:
- Direkte bilirubin > 25 μmol/L
Følgende barn skal ta en ny kontroll innen 4–7 dager:
- Direkte bilirubin 20–25 μmol/L og normalfarget avføring => ta ny total og direkte bilirubin innen 4–7 dager.
- Hvis fortsatt direkte bilirubin 20–25 μmol/L tas ny kontroll om 4–7 dager. Vurder alltid også total bilirubin og avføringsfarge. Barna bør følges til direkte bilirubin er falt under 20 μmol/L.
- Hvis stigende direkte bilirubin; vurder fenobarbital (se under) og diskusjon med barnehepatolog OUS.
Følgende barn trenger ikke videre kontroll:
- Direkte bilirubin < 20 μmol/L etter 3 uker, og klinisk frisk med normalfarget avføring (vanligvis morsmelk-assosiert gulsott)
Følgende barn vurderes individuelt med tanke på ytterligere utredning:
- Barn med svært høy total bilirubin, f.eks. nær lysgrensen eller som virker syke, uavhengig av andelen konjugert bilirubin.
Kolestase sees hyppig hos syke nyfødte og premature som er avhengig av langvarig parenteral ernæring, særlig etter tarmreseksjon.
- Vurderes separat, se IFALD under.
Flytskjema for oppfølging:
Praktisk gjennomføring langvarig gulsott
- Informasjon om forlenget gulsott: Det er ingen rutinekontroll av norske spedbarn mellom 1 og 6 ukers alder. Barselavdelingene bør dele ut infoskriv om gulsott inkl. hva man skal gjøre hvis den vedvarer. Foreldre kan selv kontakte helsestasjon/fastlege.
- Ikke vakt eller poliklinisk arbeid: Friske barn uten andre symptomer enn gulsott trenger ikke barnelegetilsyn, poliklinisk oppfølging eller å prioriteres på vakttid: Behovet er en blodprøve som svares ut elektivt.
- Vurdering av gulsott: Barn som «ser gule ut» (uavhengig av grad av gulfarge) ved 2 ukers alder hos barn som får morsmelkerstatning og ved 3 ukers alder hos barn som ammes skal ta blodprøve (total og direkte (konjugert) bilirubin.
- Organisering av blodprøvetaking, og hvem som skal stå som rekvirent, må organiseres lokalt/regionalt. Der barnet bor nært et sykehus er det antagelig enklest at blodprøven tas på sykehus. I deler av Norge der det er langt til sykehus vil det være en fordel om blodprøven tas ved noen «større» distriktmedisinske senter som kan få erfaring med denne type prøver. En kan ikke forvente at denne typen blodprøver av spedbarn kan tas ved alle fastlegekontor.
Videre utredning ved langvarig gulsott av konjugert type
Man har fokus på de tilstandene som må håndteres raskt for å unngå prognosetap:
- Korrigerbare kirurgiske tilstander: Ekstrahepatisk gallegangsatresi, koledokuscyste, obstruerende gallestein
- Infeksjon (CMV), hypofysesvikt, tyrosinemi (testes i nyfødtscreeningen), galaktosemi (kan debutere som tidlig gulsott av ukonjugert type)
Avhengig av mistenkt årsak og hva som allerede er gjort er følgende utredning aktuelt:
- Blodprøver: Hb, hvite m/diff, MCV, trombocytter, retikulocytter, ALAT, ASAT, GT, ALP, LD, CK, INR, kortisol, TSH, fritt T4, glukose, Na, K, CRP, syre-base-status, laktat, albumin, alfa-1-antitrypsin i serum.
- UL abdomen/lever/galleveier (fortrinnsvis etter 4 timers faste og etter måltid).
- Ved sykt barn vurder galaktosemi (metabolsk screening i urin) og infeksjon inkl. CMV/HSV-PCR osv. Påvisning av CMV utelukker ikke samtidig gallegangsatresi.
Nærmere informasjon om noen viktige årsaker til langvarig gulsott av konjugert type
Ekstrahepatisk gallegangsatresi
Insidens i Europa 1:18 000. Disse barna har konjugert hyperbilirubinemi, men ofte relativt upåfallende klinisk presentasjon med normal vekst og utvikling første levemåned. Mørk urin og kittfarget/hvit avføring gir sterk mistanke om diagnosen, men ses ikke hos alle.
Utredning er avhengig av lokal utredningskapasitet.
- Blodprøver: Se over
- UL lever/galleveier etter 4 timers faste og etter måltid. Tilstedeværelse av galleblære/galleblæreliknende struktur utelukker ikke ekstrahepatisk gallegangsatresi. Tegn på gallegangsatresi på ultralyd (bla. liten galleblære med uregelmessig, «knudrete» vegg, triangular cord-tegn, manglende visualisering av ductus choledochus, cyster ved leverporten, en arteria hepatica > 1,5 mm, polyspleni) kan være vanskelige å tolke. Man bør ha lav terskel for å diskutere med barnehepatolog, OUS Rikshospitalet.
- Videre utredning kan inkludere galleveisscintigrafi (inkluderer forbehandling med fenobarbital 5 mg/kg p.o. i 3–5 dager, startes ofte lokalt, men må avtales med senter der man gjør scintigrafi), perkutan transhepatisk kolangiografi og/eller leverbiopsi.
Viktig med tidlig operativ behandling, aller helst før 6–7 ukers alder.
Tarmsvikt-assosiert leversykdom (IFALD)
Kolestase sees hyppig hos nyfødte og premature som er avhengig av langvarig parenteral ernæring (PN), særlig etter tarmreseksjon. Prematuritet/intrauterin veksthemming er sterke risikofaktorer. Det er viktig å vurdere andre årsaker til kolestase (som f.eks. gallegangsobstruksjon, metabolsk sykdom, forbigående kolestase ved sepsis, medfødt eller ervervet CMV-infeksjon). Grensen for å definere patologisk konjugert hyperbilirubinemi bør være høyere (> 35–40 mikromol/L) hos barn med mistenkt IFALD enn for barn med annen årsak til sin konjugerte hyperbilirubinemi.
Enteral ernæring bør om mulig startes så fort magen er i gang etter operasjon (avgang av luft/avføring).
Parenteral ernæring bør inneholde begrenset mengde fett med en kombinasjon av soya og fiskeolje; eksempelvis SMOFlipid (inneholder både soya, MCT-fett, olivenolje og fiskeolje) 1g/kg/d og Omegaven (inneholder kun fiskeolje) 1g/kg/d.
Barn med langvarig behov for parenteral ernæring krever tverrfaglig oppfølging inklusive ernæringsfysiolog for å sikre adekvat tilførsel av både makro- og mikronutrienter.
Konferering med senter med erfaring med kirurgiske barn/langvarig PN bør vurderes.
Alfa-1-antitrypsinmangel
Alfa-1-antitrypsinmangel er en av de hyppigste autosomalt recessive arvelige sykdommer.
Over 100 ulike sekvensvarianter er observert i genet som koder for alfa-1-antitrypsin. Z- og S- er de vanligste sykdomsframkallende mutasjonene i den nordeuropeiske befolkningen, hvorav bærerfrekvensen for Z-mutasjonen er ca. 3–4 %. Homozygoti for Z-mutasjonen utgjør ca. 95 % av alle pasienter med alvorlig alfa-1-antitrypsinmangel. Pasientene kan utvikle leversykdom/kolestase med debut i nyfødtperioden eller tidlig barnealder.
Diagnosen stilles på bakgrunn av:
- Blodprøver: Alfa-1-antitrypsin i serum (obs. nyfødte har ofte normalt litt høyere verdier enn eldre barn og voksne).
- Ved lav alfa-1-antitrypsin (< 1,0 g/L) bekreftes/avkreftes diagnosen enten ved PI-typing (eller ev. genetisk testing). Slike tester tilbys ved St. Olavs hospital, OUS og Ahus.
Andre årsaker og tilhørende utredning for vedvarende gulsott hos nyfødte
- NB! Utredning og bestilling av prøver i forbindelse med antatt sjeldne årsaker til gulsott bør skje i samråd med barnelege med erfaring i kolestaseutredning. Ha lav terskel for kontakt med barnehepatolog ved OUS Rikshospitalet.
- Utredning vil ofte by på problemer relatert til prøvevolum, og prioritering av analyser må baseres på klinisk mistanke.
- For morsmelk-assosiert gulsott, ekstrahepatisk gallegangsatresi, tarmsvikt-assosiert kolestase og alfa-1-antitrypsinmangel; se egne avsnitt.
Det er sjelden/aldri grunn til å bestille alle nevnte prøver samtidig.
| Årsaker | Aktuelle prøver og undersøkelser |
| Gallegangsatresi, intrahepatisk galegangshypoplasi | Kan være ledd i syndrom.
|
| Gilbert syndrom | Kan være årsak til prolongert ukonjugert gulsott hos nyfødte med normale leverfunksjonsprøver og uten tegn til hemolyse. Imidlertid vil de fleste med Gilbert syndrom først diagnostiseres i ungdomsalder/rundt pubertet. |
| Generelle prøver, inkl. koagulasjon og elektrolytter | Ammoniakk, APTT fibrinogen, urea, urinsyre, Ca, fosfat, AFP og 25-OH vitamin D |
| Infeksjoner | Hos klinisk «syke» barn og/eller biokjemiske tegn på leversykdom med celleskade, bør infeksiøs etiologi utredes. Prøvene er aktuelle først og fremst når det foreligger klinisk mistanke om infeksiøs hepatitt og andre årsaker er lite sannsynlige/ikke påvist.
|
| Metabolsk eller endokrin sykdom | Blod: NB! Sjekk først resultatene fra nyfødtscreeningen inkl. acylkarnitiner. Diskuter prøver med spesialist. |
GALD = gestational alloimmune liver disease (tidligere kalt «Neonatal hemokromatose») | En av de vanligste, blant mange svært sjeldne, årsaker til leversvikt i nyfødtperioden. |
| Inspissated bile syndrome | Alvorlig Rh-immunisering eller andre årsaker til hemolyse, SGA/dysmature barn, syke m/redusert væskebalanse. |
| Andre tilstander | Dubin-Johnson, Rotor syndrom, sfærocytose og mistanke om andre sjeldne hemolytiske anemier. |
Tabell 1. Årsaker og tilhørende utredning for vedvarende gulsott hos nyfødte
Behandling og oppfølging
Avhengig av grunntilstand. Se lenker for relevante avsnitt eller sjekk spesiallitteratur
Ernæring og vitamintilskudd ved neonatal kolestase
Fettløselige vitaminer: Ernæring og vitamintilskudd ved neonatal kolestase
Mål plasmakonsentrasjon av fettløselige vitaminer hvis klare tegn på kolestase/leversykdom.
Følgende angis som mål under behandling:
- Serum vitamin D > 50 nmol/L
- Serum vitamin A > 0,7 μmol/L
- INR < 1,5
Hvis ikke tegn til betydelig kolestase med markert forhøyet konjugert bilirubin og forhøyet INR, eller påvist vitaminmangel, kan man initialt starte med Dekas Plus dråper 1 ml x 1 og dobbel dose vanlig vitamin D-tilskudd.
| Anbefalt mengde vitaminer (ESPGHAN 2019) < 10 kg | 1 ml Dekas Plus® Dråper + | |
| Vitamin A (μg/d) | 1500 μg/dag | 1725 μg |
| Vitamin D (μg/d) | 50–125 μg/dag | 18,8 μg + 20 μg |
| Vitamin E (mg/d) | 10–17 mg/kg/d | 33,6 mg |
| Vitamin K1 (mg) | 2–5 mg/dag | 0,5 mg |
Tabell 2. Vitamintilskudd
Ved behov og hos barn med alvorlig kolestase optimaliseres tilskudd under forløpet med forslagsvis følgende daglig dosering av fettløselige vitaminer:
- Dekas Plus® Dråper, 1 ml x 1
- Ka-vit® Dråper (20 mg/ml) 2 mg p.o.
- Detremin® 2 dråper (40 μg/dråpe) - dropper da Nycoplus® Vitamin D dråper
- Vitamin E mikstur (50 mg/ml) 40 mg x 1
Utgifter til vitamintilskudd dekkes ved refusjon §3a, individuell søknad.
Mål plasmakonsentrasjon av fettløselige vitaminer (A, D og E) underveis for å justere tilskudd under forløpet.
MCT-kost: Man kan f.eks. kombinere Lipistart (50 %) med morsmelk (50 %).
Alternativt Pregestemil (100 %). Tett samarbeid med klinisk ernæringsfysiolog.
Behandling av gallesyreindusert kløe ved kolestase
En rekke kolestatiske sykdommer har uttalt alvorlig kløe assosiert med forhøyede gallesyrer. Dette kan debutere tidlig etter fødsel eller senere i barneårene. Man bør utelukke obstruktiv kolestase i samarbeid med barnehepatolog før man starter behandling av kolestaserelatert kløe. Førstelinjebehandling vil ofte være ursodeoksykolsyre (10 mg/kg x 2–3) mikstur eller tabletter (Individuell refusjon §3a). Andre medikamenter som kan være aktuelle er kolestyramin eller rifampicin.
Referanser
- Causes of cholestasis in neonates and young infants. https://www.uptodate.com/contents/causes-of-cholestasis-in-neonates-and-young-infants
- Approach to evaluation of cholestasis in neonates and young infants og Biliary atresia og Intestinal failure-associated liver disease in infants. https://www.uptodate.com/contents/approach-to-evaluation-of-cholestasis-in-neonates-and-young-infants
- NICE Guideline: Neonatal jaundice. https://www.nice.org.uk/guidance/cg98 (25)
- Hansen TWR. Neonatal jaundice. I: Medscape eMedicine http://emedicine.medscape.com/article/974786-overview
- Kemper AR, et al. Clinical Practice Guideline Revision: Management of Hyperbilirubinemia in the Newborn Infant 35 or More Weeks of Gestation. Pediatrics. 2022;150(3): e2022058859
- Hansen TWR, et al. Molecular Physiology and Pathophysiology of Bilirubin Handling by the Blood, Liver, Intestine, and Brain in the Newborn. Physiol Rev 2020; 100:1291-346
- Bratlid D, et al. National guidelines for treatment of jaundice in the newborn. Acta Paediatr 2011; 100;499-505
- Maisels MJ, et al. The natural history of jaundice in predominantly breastfed infants. Pediatrics 2014; 134:e340-5
- Fawaz R, et al. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr 2017; 64:154-68
- Serinet MO, et al Impact of age at Kasai Operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics 2009: 123:1280-6
- Kriegermeier A, et al. Pediatric cholestatic liver disease: review of bile acid metabolism and discussion of current and emerging therapies. Front Med (Lausanne) 2020; 7:149
- Kronsten V, et al. Management of Cholestatic Pruritus in Paediatric Patients With Alagille Syndrome: The King’s College Hospital Experience. J Pediatr Gastroenterol Nutr 2013; 57:149-54
- Jaundice in early infancy. https://www.rch.org.au/clinicalguide/guideline_index/Jaundice_in_early_infancy/
- Mouzaki M, et al. Nutrition support of children with chronic liver diseases: A Joint Position Paper of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. JPGN 2019; 69:498-511.
- Ikterus. Rikshandboken Barnhalsovård, 14.09.23
https://www.rikshandboken-bhv.se/somatik/hud---oversikt/hud-nyfodda/ikterus/ - Prolongeret icterus. Dansk Pæadiatrisk selskab. Januar 2025 https://paediatri.dk/images/dokumenter/Retningslinjer_2025/Prolongeret_ikterus.pdf
- Jaundice - investigation of prolonged. 09.11.2023. Starship Clinical Guidelines New Zealand. https://www.starship.org.nz/guidelines/jaundice-investigation-of-prolonged/
Tidligere versjoner
Versjon 2023: Claus Klingenberg, Astri Lang, Thor Willy Ruud Hansen, Jannicke H Andresen, Anders Batman Mjelle, Ketil Størdal, Ingrid Nissen, Anniken Bjørnstad, Runar Almaas
Ved tilbakemeldinger eller spørsmål, send en mail til veiledere@barnelegeforeningen.no.