









Nyfødtveileder
10 Glukose, elektrolytter og metabolsk sykdom
10.7 Metabolsk sykdom med debut i nyfødtperioden
Sist faglig oppdatert: 24.04.2025
Andreas Øberg, Claus Klingenberg, Nils Thomas Songstad og Trine Tangeraas
Bakgrunn
- Enkeltvis svært sjeldne tilstander. Samlet insidens er imidlertid på 1: 2500–4000.
- Vurder metabolsk sykdom når vanlige årsaker til alvorlig sykt barn ikke forklarer forløpet (sepsis, meningitt, asfyksi, hjerneblødning, hjertesvikt, m.m.) eller ved betydelig avvikende biokjemi uten åpenbar årsak (eks. stasjonært forhøyet laktat hos «friskt» barn, grav metabolsk acidose, alvorlig hypoglykemi hos velfødd terminbarn)
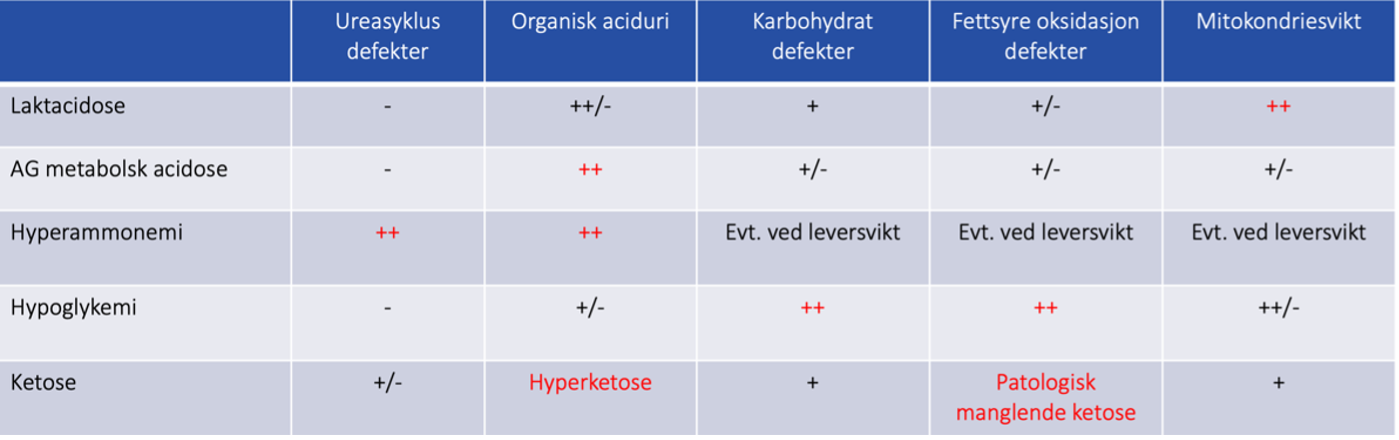
- Metabolsk sykdom fordeler seg likt hos alle nyfødte og ses derfor hyppigst hos fullbårne
- Initialt ofte uspesifikke symptomer som dårlig sugekraft, sløvhet, brekninger, irritabilitet, slapphet, takypné som ubehandlet progredierer videre til koma, apne og kramper
- Kun noen få har dysmorfe trekk (peroksisomale sykdommer, lysosomale sykdommer, og glykosyleringsdefekter)
Ureasuklusdefekter (debut fra 36 timers alder)
- Se eget avsnitt om ureasyklusdefekter.
- Ureasyklus foregår i lever og omdanner toksisk ammoniakk til urea.
- Gir encefalopatiske ammoniakkriser ved katabolsk stress. 6 undergrupper som behandles relativt likt.
- Oftest ses isolert hyperammonemi (ev. respiratorisk alkalose tidlig - og acidose sent i forløp), øvrig biokjemi kan være fredelig tross alvorlig sykt barn
Organiske acidurier (debut fra 36 timers alder)
- Forenklet 5 hovedgrupper: Maple Syrup Urine Disease (MSUD, Glutarsyreuri, Isovaleriansyreemi (IVA), Propionsyremi (PA) og Metylmalonsyreemi (MMA) - alle er inkludert i Nyfødtscreeningen, men alvorlige tilfeller blir syke før screeningsvar
- Felles: ↑Organiske syrer ved katabolsk stress +/- metabolsk acidose (opphopning av organiske syrer) +/- hyperketose +/- hypoglykemi +/- laktacidose +/- hyperammonemi
- Regn ut anion gap (AG). Metabolsk acidose med normal AG (< 20) er vanlig og skyldes oftest bikarbonattap fra tarm eller nyrer. Økt AG med normale/lett forhøyede laktatverdier gir mistanke om organisk aciduri.
- Obs.: Neonatal MSUD kan initialt manifestere seg som katastrofal encefalopati med marginal påvirkning av biokjemi (aminosyreprofil er her det eneste som gir diagnosen).
Defekter i karbohydratmetabolismen (kan debutere første levedøgn)
- Mest vanlige er glykogenlagringsdefekter. Gir hypoglykemitendens og leveraffeksjon (opphopning glykogen i lever).
Fettsyreoksidasjons defekter (kan debutere første levedøgn)
- Svikt i energiomsetning av fett og opphopning av toksiske fettsyrer.
- Neonatale varianter får rabdomyolyse +/- non-ketotisk hypoglykemi +/- kardiomyopati.
Mitokondriesykdom (kan debutere intrauterint/ved fødsel)
- Heterogen sykdomsgruppe. Ofte multiorgansykdom i form av stasjonær laktacidose (ikke obligat) samt affeksjon energikrevende organer: Retina, hjerne, lever, muskel og hjertet.
- Alvorlige varianter kan debutere intrauterint og kan dekompensere allerede ved fødsel. Typisk er lavere Apgar enn fødselsstress tilsier. MR caput med spektroskopi viser ofte laktattopper.
- Generelt svært dårlig prognose ved neonatal debut (noen få unntak)
- Ved sterk mistanke om mitokondriesykdom bes spesifikt om analyse av mitokondrielt DNA (kun nukleært DNA som analyseres i de fleste initiale genpaneler).
OBS Sykdommene kan også debutere etter nyfødtperioden, da ofte utløst av interkurrent infeksjon og/eller faste/lite næringsinntak.
Kramper i nyfødtperioden/neonatale anfall
Se eget avsnitt om neonatale anfall. Metabolsk sykdom vurderes ved kramper uten åpenbart utløsende årsak og/eller behandlingsresistente kramper.
Se også «Udredning af krampeanfald i neonatalperioden», Ugeskrift for Læger 2018 for praktisk tilnærming til metabolske kramper i nyfødtperioden:
- Pyridoksinavhengige kramper: Intraktable kramper som kan debutere in utero, kan ligne på ”HIE-barn” med funn på MR og forhøyet laktat.
- Non-ketotisk hyperglycinemi: Tidlig debut med spisevansker, alvorlig hypotoni, hikke, letargi og apneer, ev. intrauterine kramper. Progredierer raskt med myoklonier og burst suppression på EEG (senere hypsarytmi) og koma. Diagnose fås ved aminosyremåling i plasma og CSF (ratio glycin CSF/plasma), helst fra samme tidspunkt.
- Sulfitt oksidase mangel: Debut in utero eller første 1–2 leveuker med spisevansker, oppkast, hypotoni/hypertoni og intraktable kramper. Ofte mikrocefali med strukturelle CNS forandringer og ev. dysmorft ansikt. Ofte føtalt distress som kan ligne på asfyksi. Urinsyre ofte lav i serum og urin. Sulfitt testes rutinemessig med en spesialstiks. Må testes i fersk urin da sulfitt er ustabil (vil ikke alltid slå ut på metabolsk screening i urin).
- Peroksisomale sykdommer (Zellweger etc.): Migrasjonsforstyrrelser/misdannelser i CNS, alvorlig hypotoni, tidlige kramper, dysmorft ansikt, leverpåvirkning, polycystiske nyrer, skjelett- patologi. Mål ultralange fettsyrer og fytansyre i plasma.
- Glukosetransportprotein (GLUT1) mangel: Sjelden symptomdebut hos helt nyfødte, men kan debutere ila første levemåned.
- Nevrometabolske sykdommer (en del uten biomarkører)
Diagnostikk og utredning
Familieanamnese
- Uforklarlig neonatal død/sykdom (søsken, guttebarn på morsiden)? Foreldre i slekt? (de fleste sykdommer er autosomalt recessive).
Nyfødtscreening
Ved nyfødtscreeningen (2025) er følgende metabolske sykdommer inkludert:
Beta-Ketothiolasedefekt, Biotinidasedefekt, Fenylketonuri, Glutarsyreuri type 1, Glutarsyreuri type 2, Holokarbosylase syntetase defekt, Homocystinuri, Isovaleriansyreemi, Karnitin acylkarnitin translokasedefekt, Karnitin transporterdefekt, Karnitin-palmitoyltransferase 1-defekt, Karnitin-palmitoyltransferase 2-defekt, Langkjedet 3-hydroksyacyl-CoA dehydrogenasedefekt (LCHAD-defekt og Trifunksjonelt protein defekt, Maple Syrup Urine, Meget langkjedet acyl-CoA-dehydrogenasedefekt (VLCAD-defekt), Mellomkjedet acyl-CoA-dehydrogenasedefekt (MCAD-defekt), Metylmalonsyreemi (MMA), Propionsyreemi (PA), Tyrosinemi type 1 (TYR1), Ureasyklusdefektene ASS, ASL og ARG, Remetyleringsdefekter og Metakromatisk leukodystrofi.
I tillegg: Spinal muskel atrofi (SMA), sigdcelle anemi, medfødt hypotyreose, medfødt binyrebarkhyperplasi og Cystisk fibrose.
Behandlingsprotokoller for alle tilstander fins på nettsiden til Nyfødtscreeningen: https://oslo-universitetssykehus.no/avdelinger/barne-og-ungdomsklinikken/nyfodtscreeningen/nyfodtscreening
NB! Galaktosemi (kan gi akutt leversvikt i nyfødtperioden) er ikke inkludert i nyfødtscreeningen i Norge.
Standard metabolske blodprøver
Ytterligere analyser vil ofte være aktuelt senere.
- Hematologi, lever-/galle-, nyreprøver, urat, hjertemarkører, elektrolyttstatus og syre-/base
- Ketoner i blod
- Laktat: Kramper, hjertesvikt, sepsis og tarmsvikt er vanligste årsaker og kan gi verdier opptil 10–20 mmol/L. Obs. barn med mitokondriesvikt og laktacidose har ofte i tillegg akutte organkomplikasjoner (hjerte-/lever-/lungesvikt) som i seg selv kan gi laktacidose og gjør tolkning vanskelig. Kan skilles ved at laktacidoser ved mitokondriesvikt ofte bruker lenger tid på- eller ikke normaliseres etter at organfunksjon bedres.
- Pyruvat: Kun aktuelt når laktat er > 5 mmol/L for å bestemme laktat/pyruvat-ratio. Markøren brukes til å diagnostisere pyruvat dehydrogenase mangel (PDH). Høy ratio (> 20) er «normalt», lav ratio (< 10) er forenlig med PDH.
- Ammoniakk: Legges umiddelbart på is. Må tas venøst, blir falsk forhøyet ved ”skvising”
- Homocystein, MMA, folat, vit B12
- Fritt- og total karnitin (OUS-RH)
- «Metabolsk screening» sendes Medisinsk biokjemi OUS (rekvisisjon ligger på nett):
0,5 ml heparinplasma, 0,5 ml EDTA plasma og 0,5 ml serum samt 5–10 ml urin som fryses innen 45 minutter. Spinalvæske (hvis indikasjon, se under). Prøvene fryses og sendes samlet på tørris til laboratoriet og helst sammen med pasientens frosne urinprøve. Vakthavende ved medisinsk biokjemi OUS orienteres per telefon 90927517 første virkedag (prøver analyseres dagtid mandag-fredag). - EDTA-blod av barn og foreldre til genetikk.
- Nyfødtscreeningen kan kontaktes mtp. hurtiggenetikk forskningsprosjekt på Nyfødtscreeningen (tar noen få dager).
Urinprøver
- Ketoner tilstede? (ketoner i urin hos en nyfødt kan indikere medfødt stoffskiftesykdom)
- Urin til metabolsk screening inkl. aminosyrer (OUS-RH. Når det haster må begge laboratorier (lokalt/OUS-RH) varsles per telefon!)
Spinalvæske
- Celler, protein, glukose
- Relevante prøver tas parallelt i blod og CSF (glukose, laktat, pyruvat, plasma-aminosyrer).
- GLUT1 mangel: glukoseratio-CSF/plasma
- Laktat, pyruvat (blodprøve parallelt): Minst 0,5 ml, legges på is.
- Aminosyrer (OUS-RH: 0,5 ml på glass uten tilsetninger, bl.a. aktuelt ved mistanke om non-ketotisk hyperglycinemi: Glycinratio CSF/plasma. Assosiert med neonatale kramper. Lavt glycin og serin ved serinmangel.)
- Nevrotransmitter analyse i CSF sjeldent aktuelt initialt. Se hjemmeside Seksjon for medfødte metabolske sykdommer OUS for veiledning og rekvisisjon.
Relevante bildeundersøkelser ved mistanke om metabolsk sykdom
- Cerebral ultralyd, rtg. thorax, UL abdomen (hepatosplenomegali?) og ekkokardiogarfi (kardiomyopati?)
- MR caput med spektroskopi er ofte aktuelt senere i forløpet ved uavklart diagnose eller som ledd kartlegging av cerebral skade i etterkant av alvorlig metabolsk dekompensering
Postmortem leverbiopsi og hud-/hælsenebiopsi
- Fibroblaster - må tas innen 72 timer på eget transportmedium.
- Ved rask progresjon som ikke tillater sikker diagnose mens barnet var i live.
- Metabolsk sykdom var sterkt mistenkt, men testing kunne ikke påvise spesifikk defekt.
Ved spørsmål om diagnostikk/utredning vil det ofte være nyttig å rådføre seg med lege på Seksjon for biokjemisk genetikk, Med. Biokjemi, OUS-Rikshospitalet. Telefon: 23 07 10 48 eller mobil 909 27 517.
Behandling og oppfølging
NB: Se eget avsnitt om hyperammonemi og ureasyklusdefekter.
Ernæring
- Ved uavklart mistanke om metabolsk sykdom stanses proteiner (og ev. fett) i 24 timer. Proteiner kan føre til akkumulasjon av toksiske produkt (ammoniakk ved ureasyklusdefekter og organiske syrer ved organiske acidurier).
- Tilstrekkelig energi bremser katabolisme og akkumulering av toksiske metabolitter. Forgiftningstilstander (ureasyklusdefekter og organiske acidurier) krever generelt mer energi enn energisvikttilstander (karbohydrat-, fettsyreoksidasjons- og mitokondrielle defekter). Start med glukose 150 mg/ml i.v., 4,5 ml/kg/t (= 10 mg/kg/min). Tilsvarer 14,4 g/kg/d.
- Ved alvorlig sykt barn anlegges sentralvenøs tilgang
- Ved holdepunkt for fettsyreoksidasjons defekt (rabdomyolyse +/- non-ketotisk hypoglykemi) startes i.v. fett 2(-3) g/kg/d.
- Ved usikkerhet rundt kontraindikasjon for fett gis monoterapi med i.v. glukose 20 g/kg/d.
- Tilstreb 100 kcal/kg/d. Følg glukose og laktat initialt hver 1–2 time. Den høye glukosetilførselen krever ofte insulininfusjon (startdose: 0,01 E/kg/t). Målsetning er blodsukker (BS) rundt 6–10 (< 15 ofte realistisk) mmol/L. Ved mitokondriesykdom og organiske acidurier (som gir sekundær mitokondriesvikt) ses ofte intraktabel og insulinresistent hyperglykemi med behov for å gradvis redusere glukosebelastning.
- Reintroduser protein etter 24–36 timer, fortrinnsvis oralt/på sonde. Start med protein 0,3–0,5 g/kg/d (ev. som i.v. aminosyrer; Vaminolac® 0,6 g/kg/d = 0,5 g/kg/d protein). Hvis enteral ernæring ikke tolereres gis i.v. glukose, fett og aminosyrer.
- Ernæringen tilpasses grunntilstanden når diagnose er etablert.
- Ved mistanke om galaktosemi er eliminasjon av melk/melkesukker (= laktose; disakkarid av glukose og galaktose) essensielt. NB! Urinprøve må tas mens barnet får melk; prøven blir raskt falsk negativ dersom prøven tas etter melk er seponert.
Væske
- Korriger ev. væskeunderskudd. Unngå overvæsking ved encefalopati (risiko hjerneødem).
Metabolsk acidose
- Korreksjon av metabolsk acidose følger vanlige behandlingsprinsipper, dvs. vurdering av bufring ved pH < 7,10.
Infeksjon
Barn med medfødte stoffskiftesykdommer har sjelden immunsvikt. Men lav terskel for antibiotika da infeksjon forverrer metabolsk krise. Galaktosemi er assosiert med økt risiko for Gram-negativ sepsis.
Kofaktorer/vitaminbehandling
Ved kritisk syke barn hvor organisk aciduri er en mulig differensialdiagnose (eks. metabolsk acidose med forhøyet AG) gis vitamin B12 og biotin (i doser under).
Vurder følgende ved mistanke om spesifikk vitaminresponsiv sykdom:
- Vit B12: 1 mg x 1 i.m. (Hydroksykobalamin/Vit B12 Depot inj. 1 mg/ml), dag 1 og 3. Indikasjon: MMA, remetyleringsdefekter.
- Vit B7: 10 mg x 2 p.o. (Biotin kapsler a 10 mg. Kapselen åpnes og blandes i vann). Indikasjon: Biotinidasdefekt (nyfødtscreening) og Multippel karbosylasedefekt.
- Vit B6: 50 mg x 2 i.v. (Pyridoxin inj. 50 mg/ml)
- Vit B1: 100 mg x 2 i.v. (Tiamin inj. 25 mg/ml)
- Vit B2: 50 mg x 2 p.o. (Riboflavin kapsler a 50 mg. Kapselen åpnes og blandes i vann). Indikasjon: multippel Acyl-CoA dehydrogenasedefekt/Riboflavin transporter defekt
- L-karnitin gis også ved metabolsk acidose og forhøyet AG. Dose: 50 mg/kg x 4 i.v. (Carnitor® inj. 200 mg/ml) eller noe lavere dose p.o. 25–37,5 mg/kg x 4 (Biocarn® sirup 300 mg/ml). Høyere p.o. dose kan gi mageproblemer. NB! Husk å ta metabolske prøver (særlig acylkarnitiner) før karntin gis, ellers vil biokjemisk diagnose kunne maskeres.
Hemodiafiltrasjon
Ved encefalopati og mistanke om metabolsk sykdom er ved tilstander indisert med hemodiafiltrasjon (ureasyklusdefekter og MMA/PA/IVA for å dialysere ammoniakk og MSUD for å dialysere leucin). Kfr. med vakthavende lege på Nyfødt Intensiv, OUS-RH. Peritoneal dialyse er betydelig mindre effektivt og bør vanligvis IKKE benyttes med mindre barnet ikke er transportabelt. ECMO behandling kan vurderes hvis hemofiltrasjon/dialyse ikke er tilgengelig.
Forslag "Metabolsk krisepakke" - ikke tilgjengelig på alle sykehus
Pakken skal gjennomgås hver 6. måned. Skriv tydelig holdbarhetsdato.
Antall ampuller/flasker/kapsler som skal være i beredskap er angitt med fet skrift.
- L-carnitin: Biocarn® sirup 300 mg/ml (1 flaske/50 ml) - Carnitor® inj. 200 mg/ml (5 amp a 5 ml)
- Biotin: Biotin kaps. 10 mg (60 kapsler)
- Pyridoksin (vit. B6): Pyridoxin inj. 50 mg/ml (3 ampuller a 20 ml)
- Pyridoksalfosfat: Pyridoxal-5-phosphate enterotab. (Now foods) 50 mg (60 stk.)
- Kalsiumfolinat: Lederfolin tab. 5 mg (50 stk.)
- Riboflavin (vit. B2): Riboflavin kapsler (50 kapsler a 50 mg)
- Tiamin (vit. B1): Tiamin inj. 25 mg/ml (10 ampuller a 2 ml)
- Vitamin B12 Depot: Vitamin B12 Depot inj. 1 mg/ml (3 ampuller a 1 ml)
- Na-benzoat 100 mg/ml (4 glass a 50 ml)
- Argininhydroklorid (Argininklorid NAF®) 1 mmol/ml (1 glass a 100 ml)
- N-carbamylglutamat/kargluminsyre (Carbaglu®) tbl. a 200 mg, minste pakke a 5 tbl.
Ved leveringsproblemer tas først kontakt med lokalt Sykehusapotek. Alternativt Sykehusapoteket/Barneklinikken, Oslo Universitetssykehus, Rikshospitalet eller Scheele apoteket i Stockholm, tel +46 771450450 (døgnåpent, spør etter ”Metabollagret”).
Referanser
- British Inherited Metabolic Diseases Group (www.bimdg.org.uk) gir meget god informasjon om en rekke av disse sykdommene:
- Nyfødtscreeningen. https://oslo-universitetssykehus.no/avdelinger/barne-og-ungdomsklinikken/nyfodtscreeningen/nyfodtscreening
- Vernon HJ. Inborn Errors of Metabolism: Advances in Diagnosis and Therapy. JAMA Pediatr. 2015; 169:778-82.
- Mukherjee S, et al. Inborn Errors of Metabolism Screening in Neonates: Current Perspective with Diagnosis and Therapy. Curr Pediatr Rev. 2022; 18:274-85.
- Saudubray JM, et al. Inborn Errors of Metabolism Overview: Pathophysiology, Manifestations, Evaluation, and Management. Pediatr Clin North Am. 2018; 65:179-208.
Tidligere versjoner
2021: Claus Klingenberg, Nils Thomas Songstad, Trine Tangeraas